Multidimensionale Rollen von Ketonkörpern
Ketonkörper werden von der Leber erzeugt und als Energiequelle genutzt, wenn Glukose im menschlichen Körper nicht leicht verfügbar ist. Die beiden Hauptketonkörper sind Acetoacetat (AcAc) und 3-beta-Hydroxybutyrat (3HB), während Aceton der dritte und am wenigsten vorkommende Ketonkörper ist. Ketone sind immer im Blut vorhanden und ihr Spiegel steigt während des Fastens und längerer körperlicher Betätigung anKetogenese ist der biochemische Prozess, bei dem Organismen Ketonkörper durch den Abbau von Fettsäuren und ketogenen Aminosäuren herstellen.
Ketonkörper werden hauptsächlich in der Ketone erzeugt Mitochondrien von Leberzellen. Ketogenese tritt auf, wenn niedrige Blutzuckerwerte im Blut vorhanden sind, insbesondere nachdem andere zelluläre Kohlenhydratspeicher wie Glykogen erschöpft sind. Dieser Mechanismus kann auch bei unzureichenden Insulinmengen auftreten. Die Produktion von Ketonkörpern wird letztlich initiiert, um Energie bereitzustellen, die im Körper als Fettsäuren gespeichert wird. Die Ketogenese findet in den Mitochondrien statt, wo sie unabhängig reguliert wird.
Abstrakt
Der Keton-Stoffwechsel ist ein zentraler Knotenpunkt in der physiologischen Homöostase. In diesem Aufsatz diskutieren wir, wie Ketone diskreten Feinabstimmungsfunktionen dienen, die die Organ- und Organismusleistung bei verschiedenen Nährstoffresten optimieren und vor Entzündungen und Verletzungen in mehreren Organsystemen schützen. Traditionell als Stoffwechselsubstrate betrachtet, die nur bei der Kohlenhydratrestriktion eingesetzt werden, unterstreichen jüngste Beobachtungen die Bedeutung von Ketonkörpern als lebenswichtige Stoffwechsel- und Signalvermittler, wenn Kohlenhydrate im Überfluss vorhanden sind. Neben einem Repertoire bekannter therapeutischer Optionen für Erkrankungen des Nervensystems haben sich prospektive Rollen für Ketonkörper bei Krebs sowie faszinierende Schutzfunktionen in Herz und Leber ergeben und therapeutische Optionen bei mit Adipositas zusammenhängenden und kardiovaskulären Erkrankungen eröffnet. Kontroversen im Ketonstoffwechsel und in der Signalgebung werden diskutiert, um das klassische Dogma mit zeitgenössischen Beobachtungen in Einklang zu bringen.
Einleitung
Ketonkörper sind eine lebenswichtige alternative metabolische Brennstoffquelle für alle Lebensbereiche, Eukarya, Bakterien und Archaeen (Aneja et al., 2002; Cahill GF Jr, 2006; Krishnakumar et al., 2008). Der Stoffwechsel des Ketonkörpers beim Menschen wurde genutzt, um das Gehirn in episodischen Phasen des Nährstoffmangels mit Energie zu versorgen. Ketonkörper sind mit entscheidenden Stoffwechselwegen von Säugetieren wie der &bgr;-Oxidation (FAO), dem Tricarbonsäurezyklus (TCA), der Gluconeogenese, der De-novo-Lipogenese (DNL) und der Biosynthese von Sterolen verwoben. Bei Säugetieren werden Ketonkörper überwiegend in der Leber aus FAO-abgeleitetem Acetyl-CoA produziert und zur terminalen Oxidation in extrahepatisches Gewebe transportiert. Diese Physiologie liefert einen alternativen Brennstoff, der durch relativ kurze Fastenperioden ergänzt wird, was die Verfügbarkeit von Fettsäuren erhöht und die Verfügbarkeit von Kohlenhydraten verringert (Cahill GF Jr, 2006; McGarry und Foster, 1980; Robinson und Williamson, 1980). Die Ketonkörperoxidation trägt in einer Vielzahl von physiologischen Zuständen, einschließlich Fasten, Hungern, Neugeborenenperiode, Post-Sport, Schwangerschaft und Einhaltung einer kohlenhydratarmen Diät, zu einem signifikanten Beitrag zum gesamten Energiestoffwechsel von Säugetieren in extrahepatischen Geweben bei. Die zirkulierenden Gesamtketonkörperkonzentrationen bei gesunden erwachsenen Menschen zeigen normalerweise zirkadiane Oszillationen zwischen ca. Cahill GF Jr, 100; Johnson et al., 250b; Koeslag et al., 1; Robinson und Williamson, 24; Wildenhoff et al., 20). Die menschliche Leber produziert bis zu 2006 g Ketonkörper pro Tag (Balasse und Fery, 1969), die zwischen 1980 % des gesamten Energieverbrauchs in genährten, nüchternen und verhungerten Zuständen beitragen (Balasse et al., 1980; Cox et al., 1974).
Kürzlich durchgeführte Studien zeigen, wie wichtig die Rolle von Ketonkörpern beim Stoffwechsel der Säugerzellen, bei der Homöostase und bei der Signalgebung unter einer Vielzahl physiologischer und pathologischer Zustände ist. Ketonkörper dienen nicht nur als Energieträger für extrahepatische Gewebe wie Gehirn, Herz oder Skelettmuskel, sie spielen auch eine entscheidende Rolle als Signalvermittler, Treiber der posttranslationalen Modifikation von Proteinen (PTM) und Modulatoren für Entzündungen und oxidativen Stress. In dieser Übersicht geben wir sowohl klassische als auch moderne Ansichten über die pleiotropen Rollen von Ketonkörpern und deren Stoffwechsel.
Überblick über den Ketone-Körperstoffwechsel
Die Geschwindigkeit der hepatischen Ketogenese wird durch eine orchestrierte Reihe physiologischer und biochemischer Transformationen von Fett bestimmt. Zu den primären Regulatoren gehören die Lipolyse von Fettsäuren aus Triacylglycerinen, der Transport zur und über die Hepatozyten-Plasmamembran, der Transport in Mitochondrien über Carnitin-Palmitoyltransferase 1 (CPT1), die & agr; -Oxidationsspirale, die TCA-Zyklusaktivität und Zwischenkonzentrationen, das Redoxpotential und die hormonellen Regulatoren von diesen Prozessen vorwiegend Glucagon und Insulin [Übersicht in (Arias et al., 1995; Ayte et al., 1993; Ehara et al., 2015; Ferre et al., 1983; Kahn et al., 2005; McGarry und Foster) 1980; Williamson et al., 1969)]. Die klassische Ketogenese wird als Spillover-Weg angesehen, bei dem von & agr; -oxidation abgeleitetes Acetyl-CoA die Citrat-Synthase-Aktivität und / oder die Verfügbarkeit von Oxalacetat für die Kondensation zur Bildung von Citrat übersteigt. Drei-Kohlenstoff-Zwischenprodukte zeigen eine antiketogene Aktivität, vermutlich aufgrund ihrer Fähigkeit, den Oxalacetat-Pool für den Acetyl-CoA-Verbrauch zu erweitern, aber die Acetyl-CoA-Konzentration in der Leber allein bestimmt nicht die ketogene Rate (Foster, 1967; Rawat und Menahan, 1975; Williamson) et al., 1969). Die Regulation der Ketogenese durch hormonelle, transkriptionelle und posttranslationale Ereignisse zusammen stützen die Annahme, dass die molekularen Mechanismen zur Feinabstimmung der ketogenen Rate unvollständig verstanden bleiben (siehe Regulation von HMGCS2 und SCOT / OXCT1).
Die Ketogenese findet hauptsächlich in der mitochondrialen Lebermatrix mit Raten statt, die proportional zur Gesamtfettoxidation sind. Nach dem Transport von Acylketten durch die Mitochondrienmembranen und der & agr; -Oxidation katalysiert die mitochondriale Isoform der 3-Hydroxymethylglutaryl-CoA-Synthase (HMGCS2) das Schicksal, das die Kondensation von Acetoacetyl-CoA (AcAc-CoA) und Acetyl-CoA zur Erzeugung von HMG-CoA bewirkt (Fig. 1A). HMG-CoA-Lyase (HMGCL) spaltet HMG-CoA unter Freisetzung von Acetyl-CoA und Acetoacetat (AcAc), und letzteres wird durch Phosphatidylcholin-abhängige mitochondriale d- & OHgr; OHB-Dehydrogenase (d- & agr; OHB) zu d- & agr; -Hydroxybutyrat (d- & agr; OHB) reduziert BDH1) in einer NAD + / NADH-gekoppelten Nahgleichgewichtsreaktion (Bock und Fleischer, 1975; LEHNINGER et al., 1960). Die BDH1-Gleichgewichtskonstante begünstigt die d-OHB-Produktion, aber das Verhältnis von AcAc / d-OHB-Ketonkörpern ist direkt proportional zum mitochondrialen NAD + / NADH-Verhältnis, und somit moduliert die BDH1-Oxidoreduktaseaktivität das mitochondriale Redoxpotential (Krebs et al., 1969; Williamson et al., 1967). AcAc kann auch spontan zu Aceton decarboxylieren (Pedersen, 1929), der Quelle für süßen Geruch bei Menschen mit Ketoazidose (dh Gesamtserumketonkörper> ~ 7 mM; AcAc pKa 3.6, OHB pKa 4.7). Die Mechanismen, durch die Ketonkörper durch die mitochondriale Innenmembran transportiert werden, sind nicht bekannt, aber AcAc / d- & OHgr; OHB werden über Monocarboxylattransporter aus Zellen freigesetzt (bei Säugetieren MCT 1 und 2, auch bekannt als Mitglieder 16 und 1 der Familie der gelösten Träger 7A) 2011) und im Kreislauf zu extrahepatischen Geweben zur terminalen Oxidation transportiert (Cotter et al., 2012; Halestrap und Wilson, 2012; Halestrap, 2012; Hugo et al., 1940). Die Konzentrationen von zirkulierenden Ketonkörpern sind höher als die in den extrahepatischen Geweben (Harrison und Long, 1), was darauf hinweist, dass Ketonkörper über einen Konzentrationsgradienten transportiert werden. Funktionsverlustmutationen in MCTXNUMX sind mit spontanen Ketoazidose-Anfällen verbunden, was auf eine entscheidende Rolle beim Import von Ketonkörpern hinweist.

Mit Ausnahme einer möglichen Umlenkung von Ketonkörpern in nicht-oxidative Schicksale (siehe Nicht-oxidative Stoffwechselschicksale von Ketonkörpern) fehlt den Hepatozyten die Fähigkeit, die von ihnen produzierten Ketonkörper zu metabolisieren. Ketonkörper, die de novo von der Leber synthetisiert werden, werden (i) in Mitochondrien extrahepatischer Gewebe zu Acetyl-CoA katabolisiert, das dem TCA-Zyklus für die terminale Oxidation zur Verfügung steht (Abb. 1A), (ii) auf die Lipogenese- oder Sterolsynthesewege umgeleitet ( Abb. 1B) oder (iii) mit dem Urin ausgeschieden. Als alternativer energetischer Brennstoff werden Ketonkörper im Herzen, in der Skelettmuskulatur und im Gehirn eifrig oxidiert (Balasse und Fery, 1989; Bentourkia et al., 2009; Owen et al., 1967; Reichard et al., 1974; Sultan, 1988 ). Extrahepatisches mitochondriales BDH1 katalysiert die erste Reaktion der &agr;OHB-Oxidation und wandelt es in rückseitiges AcAc um (LEHNINGER et al., 1960; Sandermann et al., 1986). Eine zytoplasmatische d-?OHB-Dehydrogenase (BDH2) mit nur 20% Sequenzidentität zu BDH1 hat einen hohen Km für Ketonkörper und spielt auch eine Rolle bei der Eisenhomöostase (Davuluri et al., 2016; Guo et al., 2006) . In der extrahepatischen mitochondrialen Matrix wird AcAc zu AcAc-CoA durch Austausch einer CoA-Einheit von Succinyl-CoA in einer Reaktion aktiviert, die durch eine einzigartige Säugetier-CoA-Transferase, Succinyl-CoA:3-Oxosäure-CoA-Transferase (SCOT, CoA-Transferase; kodiert durch OXCT1), durch eine nahezu Gleichgewichtsreaktion. Die durch Hydrolyse von AcAc-CoA freigesetzte freie Energie ist größer als die von Succinyl-CoA, was die AcAc-Bildung begünstigt. Somit tritt der oxidative Fluss des Ketonkörpers aufgrund der Massenwirkung auf: eine reichliche Zufuhr von AcAc und ein schneller Verbrauch von Acetyl-CoA durch die Citratsynthase begünstigt die Bildung von AcAc-CoA (+ Succinat) durch SCOT. Im Gegensatz zu Glucose (Hexokinase) und Fettsäuren (Acyl-CoA-Synthetasen) erfordert die Aktivierung von Ketonkörpern (SCOT) in eine oxidierbare Form keine Investition von ATP. Eine reversible AcAc-CoA-Thiolase-Reaktion [katalysiert durch eine der vier mitochondrialen Thiolasen, die entweder von ACAA2 (die ein Enzym namens T1 oder CT kodiert), ACAT1 (das T2 kodiert), HADHA oder HADHB kodiert, ergibt zwei Moleküle Acetyl-CoA, die in den TCA-Zyklus eintreten (Hersh und Jencks, 1967; Stern et al., 1956; Williamson et al., 1971). Während ketotischer Zustände (dh Gesamtserumketone > 500 µM) tragen Ketonkörper erheblich zum Energieverbrauch bei und werden im Gewebe schnell verwertet, bis eine Aufnahme oder Oxidationssättigung eintritt (Balasse et al., 1978; Balasse und Fery, 1989) ; Edmond et al., 1987). Ein sehr kleiner Anteil der aus der Leber stammenden Ketonkörper kann leicht im Urin gemessen werden, und die Verwertungs- und Resorptionsraten durch die Niere sind proportional zur zirkulierenden Konzentration (Goldstein, 1987; Robinson und Williamson, 1980). Während stark ketotischer Zustände (> 1 mM im Plasma) dient Ketonurie als halbquantitativer Reporter der Ketose, obwohl die meisten klinischen Assays von Ketonkörpern im Urin AcAc, aber nicht ?OHB nachweisen (Klocker et al., 2013).
Ketogene Substrate und ihre Auswirkungen auf den Hepatozytenstoffwechsel
Ketogene Substrate umfassen Fettsäuren und Aminosäuren (Fig. 1B). Der Katabolismus von Aminosäuren, insbesondere von Leucin, erzeugt etwa 4% Ketonkörper im postabsorptiven Zustand (Thomas et al., 1982). Der Acetyl-CoA-Substratpool zur Erzeugung von Ketonkörpern stammt daher hauptsächlich von Fettsäuren, da Pyruvat während Zuständen mit verminderter Kohlenhydratzufuhr hauptsächlich durch Anaplerose, dh durch ATP-abhängige Carboxylierung zu Oxaloacetat (OAA) oder zu Malat, in den hepatischen TCA-Zyklus eintritt (MAL) und keine oxidative Decarboxylierung zu Acetyl-CoA (Jeoung et al., 2012; Magnusson et al., 1991; Merritt et al., 2011). In der Leber tragen Glucose und Pyruvat vernachlässigbar zur Ketogenese bei, selbst wenn die Decarboxylierung von Pyruvat zu Acetyl-CoA maximal ist (Jeoung et al., 2012).
Acetyl-CoA nimmt verschiedene Rollen ein, die für den hepatischen Intermediärstoffwechsel über die ATP-Erzeugung durch terminale Oxidation wesentlich sind (siehe auch Die Integration des Keton-Körperstoffwechsels, die posttranslationale Modifikation und die Zellphysiologie). Acetyl-CoA aktiviert allosterisch (i) Pyruvatcarboxylase (PC) und aktiviert dadurch einen metabolischen Kontrollmechanismus, der einen hefterotischen Eintritt von Metaboliten in den TCA-Zyklus (Owen et al., 2002; Scrutton und Utter, 1967) und (ii) Pyruvatdehydrogenase verstärkt Kinase, die Pyruvatdehydrogenase (PDH) phosphoryliert und inhibiert (Cooper et al., 1975), wodurch der Pyruvatfluss durch Anaplerose in den TCA-Zyklus weiter verbessert wird. Cytoplasmatisches Acetyl-CoA, dessen Pool durch Mechanismen, die mitochondriales Acetyl-CoA zu transportierbaren Metaboliten umwandeln, verstärkt wird, hemmt die Fettsäure-Oxidation: Acetyl-CoA-Carboxylase (ACC) katalysiert die Umwandlung von Acetyl-CoA zu Malonyl-CoA, dem lipogenen Substrat und allosterischer Inhibitor von mitochondrialem CPT1 [im Überblick (Kahn et al., 2005; McGarry und Foster, 1980)]. Somit reguliert der mitochondriale Acetyl-CoA-Pool sowohl den Spillover-Weg der Ketogenese, der die zentralen Aspekte des Intermediärstoffwechsels der Leber steuert.
Nichtoxidative metabolische Schicksale von Ketonkörpern
Das vorherrschende Schicksal von aus der Leber stammenden Ketonen ist die SCOT-abhängige extrahepatische Oxidation. AcAc kann jedoch aus Mitochondrien exportiert und auf anabolen Wegen durch Umwandlung in AcAc-CoA durch eine ATP-abhängige Reaktion verwendet werden, die durch cytoplasmatische Acetoacetyl-CoA-Synthetase (AACS, Abb. 1B) katalysiert wird. Dieser Weg ist während der Entwicklung des Gehirns und in der laktierenden Brustdrüse aktiv (Morris, 2005; Robinson und Williamson, 1978; Ohgami et al., 2003). AACS wird auch in Fettgewebe und aktivierten Osteoklasten stark exprimiert (Aguilo et al., 2010; Yamasaki et al., 2016). Cytoplasmatisches AcAc-CoA kann entweder durch cytosolisches HMGCS1 auf die Sterolbiosynthese gerichtet sein oder durch eine der beiden cytoplasmatischen Thiolasen zu Acetyl-CoA (ACAA1 und ACAT2) gespalten, zu Malonyl-CoA carboxyliert werden und zur Synthese von Fettsäuren beitragen al., 1984; Edmond, 1974; Endemann et al., 1982; Geelen et al., 1983; Webber und Edmond, 1977).
Während die physiologische Bedeutung noch nicht geklärt ist, können Ketone sogar in der Leber als anabole Substrate dienen. In künstlichen experimentellen Kontexten kann AcAc bis zur Hälfte des neu synthetisierten Lipids und bis zu 75% des neu synthetisierten Cholesterins beitragen (Endemann et al., 1982; Geelen et al., 1983; Freed et al., 1988). Da AcAc aus einer unvollständigen Fettoxidation in der Leber stammt, würde die Fähigkeit von AcAc, in vivo zur Lipogenese beizutragen, einen sinnlosen Zyklus der Leber implizieren, bei dem aus Fett abgeleitete Ketone für die Lipidproduktion verwendet werden können, eine Vorstellung, deren physiologische Bedeutung experimentelle Validierung erfordert, aber dienen könnte adaptive oder maladaptive Rollen (Solinas et al., 2015). AcAc liefert eifrig die Cholesterogenese, wobei ein niedriger AACS-Km-AcAc (~50 µM) die AcAc-Aktivierung sogar im gefütterten Zustand begünstigt (Bergstrom et al., 1984). Die dynamische Rolle des zytoplasmatischen Ketonstoffwechsels wurde in primären embryonalen Neuronen der Maus und in von 3T3-L1 abgeleiteten Adipozyten vorgeschlagen, da der AACS-Knockdown die Differenzierung jedes Zelltyps beeinträchtigt (Hasegawa et al., 2012a; Hasegawa et al., 2012b). Knockdown von AACS bei Mäusen in vivo senkte das Serumcholesterin (Hasegawa et al., 2012c). SREBP-2, ein Master-Transkriptionsregulator der Cholesterinbiosynthese und Peroxisom-Proliferator-aktivierter Rezeptor (PPAR)-&bgr; sind AACS-Transkriptionsaktivatoren und regulieren ihre Transkription während der Neuritenentwicklung und in der Leber (Aguilo et al., 2010; Hasegawa et al., 2012c). Zusammengefasst kann der zytoplasmatische Ketonkörpermetabolismus bei ausgewählten Zuständen oder Krankheitsverlauf wichtig sein, ist jedoch nicht ausreichend, um aus der Leber stammende Ketonkörper zu beseitigen, da eine massive Hyperketonämie im Rahmen einer selektiven Beeinträchtigung des primären oxidativen Schicksals durch Funktionsverlustmutationen auftritt zu SCOT (Berry et al., 2001; Cotter et al., 2011).
Regulierung von HMGCS2 und SCOT / OXCT1
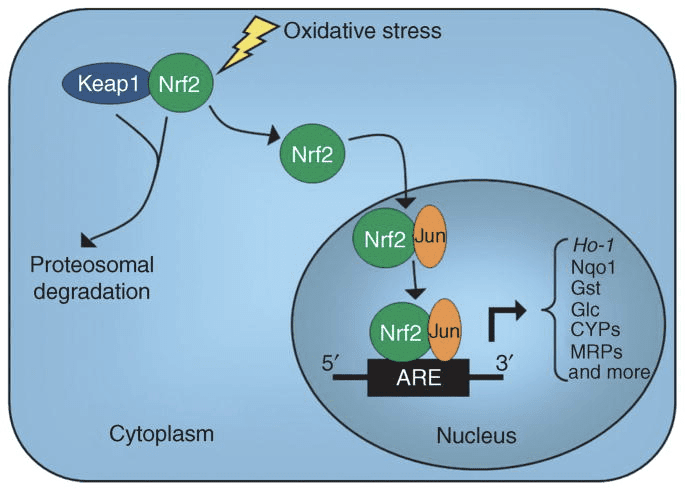
Die Divergenz eines Mitochondriums von dem Gen, das für zytosolisches HMGCS kodiert, trat früh in der Wirbeltierevolution auf, weil die hepatische Ketogenese bei Arten mit höheren Verhältnissen von Gehirn zu Körpergewicht unterstützt werden muss (Boukaftane et al., 1994; Cunnane und Crawford, 2003). Natürlich vorkommende HMGCS2-Mutationen mit Funktionsverlust beim Menschen verursachen Anfälle von hypoketotischer Hypoglykämie (Pitt et al., 2015; Thompson et al., 1997). Die robuste HMGCS2-Expression ist auf Hepatozyten und Dickdarmepithel beschränkt und ihre Expression und enzymatische Aktivität werden durch verschiedene Mechanismen koordiniert (Mascaro et al., 1995; McGarry und Foster, 1980; Robinson und Williamson, 1980). Während der volle Umfang physiologischer Zustände, die HMGCS2 beeinflussen, einer weiteren Aufklärung bedarf, ist seine Expression und / oder Aktivität während der frühen postnatalen Phase, Alterung, Diabetes, Hunger oder Einnahme ketogener Ernährung (Balasse und Fery, 1989; Cahill GF Jr, 2006) reguliert Girard et al., 1992; Hegardt, 1999; Satapati et al., 2012; Sengupta et al., 2010). Beim Fötus korreliert die Methylierung der 5. flankierenden Region des Hmgcs2-Gens umgekehrt mit seiner Transkription und wird nach der Geburt teilweise rückgängig gemacht (Arias et al., 1995; Ayte et al., 1993; Ehara et al., 2015; Ferre et al ., 1983). In ähnlicher Weise zeigt hepatisches Bdh1 ein Entwicklungs-Expressionsmuster, das von Geburt an bis zum Absetzen zunimmt, und wird auch durch ketogene Diät in einer Fibroblasten-Wachstumsfaktor (FGF) -21-abhängigen Weise induziert (Badman et al., 2007; Zhang et al., 1989 ). Die Ketogenese bei Säugetieren reagiert sehr stark auf Insulin und Glucagon und wird unterdrückt bzw. stimuliert (McGarry und Foster, 1977). Insulin unterdrückt die Fettgewebe des Fettgewebes, wodurch die Ketogenese des Substrats unterbunden wird, während Glucagon den ketogenen Fluss durch direkte Wirkung auf die Leber erhöht (Hegardt, 1999). Die Transkription von Hmgcs2 wird durch den Forkhead-Transkriptionsfaktor FOXA2 stimuliert, der durch Insulinphosphatidylinosit-3-Kinase / Akt inhibiert wird, und wird durch Glucagon-cAMP-p300-Signalisierung (Arias et al., 1995; Hegardt, 1999; Qu. Et al.) Induziert. 1990; Thumelin et al., 1993; von Meyenn et al., 2013; Wolfrum et al., 2004; Wolfrum et al., 2003). PPAR? (Rodriguez et al., 1994) induzieren zusammen mit seinem Ziel FGF21 (Badman et al., 2007) auch die Hmgcs2-Transkription in der Leber während des Hungerns oder der Verabreichung einer ketogenen Diät (Badman et al., 2007; Inagaki et al., 2007) ). Induktion von PPAR? kann vor dem Übergang von der fetalen zur neonatalen Physiologie auftreten, während die Aktivierung von FGF21 in der frühen Neugeborenenperiode durch & Dgr; OHB-vermittelte Hemmung der Histondeacetylase (HDAC) -3 begünstigt werden kann (Rando et al., 2016). mTORC1 (Säugetierziel von Rapamycinkomplex 1) abhängige Hemmung von PPAR? Die Transkriptionsaktivität ist auch ein Schlüsselregulator der Hmgcs2-Genexpression (Sengupta et al., 2010), und Leber-PER2, ein zirkadianer Master-Oszillator, reguliert indirekt die Hmgcs2-Expression (Chavan et al., 2016). Jüngste Beobachtungen deuten darauf hin, dass extrahepatisches Tumor-induziertes Interleukin-6 die Ketogenese über PPAR beeinträchtigt. Unterdrückung (Flint et al., 2016).
Die HMGCS2-Enzymaktivität wird durch mehrere PTMs reguliert. Die Serumphosphorylierung von HMGCS2 erhöhte seine Aktivität in vitro (Grimsrud et al., 2012). Die HMGCS2-Aktivität wird allosterisch durch Succinylierung von Succinyl-CoA und Lysinrest gehemmt (Arias et al., 1995; Hegardt, 1999; Lowe und Tubbs, 1985; Quant et al., 1990; Rardin et al., 2013; Reed et al. 1975; Thumelin et al., 1993). Die Succinylierung von HMGCS2-, HMGCL- und BDH1-Lysinresten in hepatischen Mitochondrien sind Ziele des NAD + -abhängigen Deacylase-Sirtuins 5 (SIRT5) (Rardin et al., 2013). Die HMGCS2-Aktivität wird auch durch SIRT3-Lysin-Deacetylierung verstärkt, und es ist möglich, dass ein Übersprechen zwischen Acetylierung und Succinylierung die HMGCS2-Aktivität reguliert (Rardin et al., 2013; Shimazu et al., 2013). Trotz der Fähigkeit dieser PTMs, HMGCS2 Km und Vmax zu regulieren, wurden Schwankungen dieser PTMs noch nicht sorgfältig kartiert und als mechanistische Treiber der Ketogenese in vivo nicht bestätigt.
SCOT wird in allen Säugerzellen, die Mitochondrien beherbergen, mit Ausnahme der Hepatozyten, exprimiert. Die Bedeutung der SCOT-Aktivität und der Ketolyse wurde in SCOT-KO-Mäusen gezeigt, die eine einheitliche Letalität aufgrund einer hyperketonämischen Hypoglykämie innerhalb von 48h nach der Geburt aufwiesen (Cotter et al., 2011). Gewebespezifischer SCOT-Verlust in Neuronen oder Skelettmyozyten induziert Stoffwechselanomalien während des Hungers, ist jedoch nicht tödlich (Cotter et al., 2013b). Beim Menschen ist der SCOT-Mangel schon früh mit schwerer Ketoazidose verbunden, was zu Lethargie, Erbrechen und Koma führt (Berry et al., 2001; Fukao et al., 2000; Kassovska-Bratinova et al., 1996; Niezen-Koning et al. 1997; Saudubray et al., 1987; Snyderman et al., 1998; Tildon und Cornblath, 1972). Auf zellulärer Ebene ist relativ wenig über SCOT-Gen- und Proteinexpressionsregulatoren bekannt. Oxct1-mRNA-Expression und SCOT-Protein und -Aktivität werden in ketotischen Zuständen möglicherweise durch PPAR-abhängige Mechanismen vermindert (Fenselau und Wallis, 1974; Fenselau und Wallis, 1976; Grinblat et al., 1986; Okuda et al., 1991; Turko et al 2001; Wentz et al., 2010). Bei diabetischer Ketoazidose wird das Missverhältnis zwischen hepatischer Ketogenese und extrahepatischer Oxidation durch die Beeinträchtigung der SCOT-Aktivität verstärkt. Die Überexpression des Insulin-unabhängigen Glucosetransporters (GLUT1 / SLC2A1) in Kardiomyozyten inhibiert auch die Oxct1-Genexpression und reguliert die terminale Oxidation der Ketone in einem nicht-ketotischen Zustand (Yan et al., 2009). In der Leber wird die Oxct1-mRNA-Abundanz durch microRNA-122 und die Histon-Methylierung H3K27me3, die während des Übergangs von der fötalen in die neonatale Periode sichtbar werden, unterdrückt (Thorrez et al., 2011). Die Unterdrückung der hepatischen Oxct1-Expression in der postnatalen Periode ist jedoch in erster Linie auf die Evakuierung von Oxct1-exprimierenden hämatopoetischen Vorläuferzellen aus der Leber zurückzuführen, und nicht auf den Verlust der zuvor vorhandenen Oxct1-Expression in terminal differenzierten Hepatozyten. Tatsächlich ist die Expression von Oxct1-mRNA und SCOT-Protein in differenzierten Hepatozyten extrem niedrig (Orii et al., 2008).
SCOT wird auch von PTMs reguliert. Das Enzym ist im Gehirn von SIRT3-KO-Mäusen hyperacetyliert, die auch eine verminderte AcAc-abhängige Acetyl-CoA-Produktion aufweisen (Dittenhafer-Reed et al., 2015). Die nicht-enzymatische Nitrierung von Tyrosinresten von SCOT schwächt ebenfalls seine Aktivität ab, was in Herzen verschiedener diabetischer Mausmodelle berichtet wurde (Marcondes et al., 2001; Turko et al., 2001; Wang et al., 2010a). Im Gegensatz dazu steigert die Nitrierung von Tryptophanresten die SCOT-Aktivität (Br g re et al., 2010; Rebrin et al., 2007). Molekulare Mechanismen der rückstandsspezifischen Nitrierung oder Denitrierung, die darauf abzielen, die SCOT-Aktivität zu modulieren, können existieren und bedürfen der Aufklärung.
Kontroversen in der extrahepatischen Ketogenese
Bei Säugetieren ist das primäre ketogene Organ die Leber, und nur Hepatozyten und Darmepithelzellen exprimieren reichlich die mitochondriale Isoform von HMGCS2 (Cotter et al., 2013a; Cotter et al., 2014; McGarry und Foster, 1980; Robinson und Williamson, 1980). . Die anaerobe bakterielle Fermentation komplexer Polysaccharide ergibt Butyrat, das von Kolonozyten in Säugetieren zur terminalen Oxidation oder Ketogenese absorbiert wird (Cherbuy et al., 1995), das eine Rolle bei der Kolonozytendifferenzierung spielen könnte (Wang et al., 2016). Mit Ausnahme von Darmepithelzellen und Hepatozyten ist HMGCS2 in fast allen anderen Säugerzellen fast nicht vorhanden, aber die Aussicht auf eine extrahepatische Ketogenese wurde in Tumorzellen, Astrozyten des zentralen Nervensystems, der Niere, der Bauchspeicheldrüse ? Zellen, retinalem Pigmentepithel (RPE) und sogar im Skelettmuskel (Adijanto et al., 2014; Avogaro et al., 1992; El Azzouny et al., 2016; Grabacka et al., 2016; Kang et al., 2015 ; Le Foll et al., 2014; Nonaka et al., 2016; Takagi et al., 2016a; Thevenet et al., 2016; Zhang et al., 2011). Ektopisches HMGCS2 wurde in Geweben beobachtet, denen die Netto-Ketogenkapazität fehlt (Cook et al., 2016; Wentz et al., 2010) , 2; Kostiuk et al., 2016; Meertens et al., 2010).
Jedes extrahepatische Gewebe, das Ketonkörper oxidiert, kann auch Ketonkörper über HMGCS2-unabhängige Mechanismen akkumulieren (Abb. 2A). Es gibt jedoch kein extrahepatisches Gewebe, in dem eine stationäre Ketonkörperkonzentration die im Kreislauf übersteigt (Cotter et al., 2011; Cotter et al., 2013b; Harrison und Long, 1940), was unterstreicht, dass Ketonkörper nach a transportiert werden Konzentrationsgradient über MCT1 / 2-abhängige Mechanismen. Ein Mechanismus der offensichtlichen extrahepatischen Ketogenese kann tatsächlich eine relative Beeinträchtigung der Ketonoxidation widerspiegeln. Zusätzliche mögliche Erklärungen fallen in den Bereich der Ketonkörperbildung. Erstens kann die De-novo-Ketogenese über die reversible enzymatische Aktivität von Thiolase und SCOT erfolgen (Weidemann und Krebs, 1969). Wenn die Konzentration von Acetyl-CoA relativ hoch ist, laufen Reaktionen, die normalerweise für die AcAc-Oxidation verantwortlich sind, in umgekehrter Richtung ab (GOLDMAN, 1954). Ein zweiter Mechanismus tritt auf, wenn sich von der & agr; -Oxidation abgeleitete Zwischenprodukte aufgrund eines TCA-Zyklus-Engpasses ansammeln. AcAc-CoA wird durch eine durch mitochondriale 3-Hydroxyacyl-CoA-Dehydrogenase und weiter durch 3-Hydroxybutyryl katalysierte Reaktion in 1980- & OHgr; OHB-CoA umgewandelt CoA-Deacylase zu 2011-OHB, die durch Massenspektrometrie oder Resonanzspektroskopie nicht vom physiologischen Enantiomer d-OHB zu unterscheiden ist (Reed und Ozand, 1984). l-OHB kann chromatographisch oder enzymatisch von d-OHB unterschieden werden und ist in extrahepatischen Geweben vorhanden, jedoch nicht in Leber oder Blut (Hsu et al., 1987). Die hepatische Ketogenese erzeugt nur d-OHB, das einzige Enantiomer, das ein BDH-Substrat ist (Ito et al., 1980; Lincoln et al., 1982; Reed und Ozand, 1982; Scofield et al., 2; Scofield et al., 1990). Ein dritter HMGCS1988-unabhängiger Mechanismus erzeugt d-OHB durch Aminosäurekatabolismus, insbesondere den von Leucin und Lysin. Ein vierter Mechanismus ist nur offensichtlich, weil er auf ein Markierungsartefakt zurückzuführen ist und daher als Pseudoketogenese bezeichnet wird. Dieses Phänomen ist auf die Reversibilität der SCOT- und Thiolase-Reaktionen zurückzuführen und kann aufgrund der Isotopenverdünnung des Ketonkörper-Tracers in extrahepatischem Gewebe zu einer Überschätzung des Ketonkörperumsatzes führen (Des Rosiers et al., 1990; Fink et al., 1978). . Dennoch kann die Pseudoketogenese in den meisten Zusammenhängen vernachlässigbar sein (Bailey et al., 2; Keller et al., XNUMX). Ein Schema (Fig. XNUMXA) zeigt einen nützlichen Ansatz zur Anwendung unter Berücksichtigung einer erhöhten Konzentration von Ketonen im stationären Gewebe.


Die Niere hat in letzter Zeit als potenziell ketogenes Organ Aufmerksamkeit erregt. In der überwiegenden Mehrheit der Staaten ist die Niere ein Nettoverbraucher von Ketonkörpern aus der Leber, die Ketonkörper aus dem Blutkreislauf ausscheiden oder resorbieren, und die Niere ist im Allgemeinen kein Netto-Ketonkörpergenerator oder -konzentrator (Robinson und Williamson, 1980). Die Autoren einer klassischen Studie kamen zu dem Schluss, dass die in einem künstlichen experimentellen System quantifizierte minimale renale Ketogenese physiologisch nicht relevant ist (Weidemann und Krebs, 1969). Kürzlich wurde in diabetischen und autophagiedefizienten Mausmodellen auf eine renale Ketogenese geschlossen, aber es ist wahrscheinlicher, dass Multiorganverschiebungen in der metabolischen Homöostase den integrativen Ketonstoffwechsel durch Eingaben auf mehrere Organe verändern (Takagi et al., 2016a; Takagi et al., 2016b; Zhang et al., 2011). Eine kürzlich veröffentlichte Veröffentlichung schlug die renale Ketogenese als Schutzmechanismus gegen Ischämie-Reperfusionsschäden in der Niere vor (Tran et al., 2016). Absolute Steady-State-Konzentrationen von &agr;OHB aus Extrakten von Nierengewebe von Mäusen wurden mit ~4 mM angegeben. Um zu testen, ob dies haltbar war, quantifizierten wir die ?OHB-Konzentrationen in Nierenextrakten von gefütterten und 12 Stunden nüchternen Mäusen. Die Serum-&bgr;OHB-Konzentrationen stiegen von ~24 µM auf 100 mM beim 2-Stunden-Fasten (Abb. 24C E), Beobachtungen, die mit Konzentrationen übereinstimmen, die vor über 2 Jahren quantifiziert wurden (Hems und Brosnan, 100). Es bleibt möglich, dass in ketotischen Zuständen aus der Leber stammende Ketonkörper renoprotektiv sein könnten, aber der Beweis für eine renale Ketogenese erfordert eine weitere Begründung. In RPE wurden überzeugende Beweise vorgelegt, die eine echte extrahepatische Ketogenese unterstützen (Adijanto et al., 1). Diese faszinierende metabolische Transformation wurde vorgeschlagen, um möglicherweise den Fluss von RPE-abgeleiteten Ketonen zu Photorezeptor- oder Müller-Glia-Zellen zu ermöglichen, was bei der Regeneration des äußeren Photorezeptorsegments helfen könnte.
OHB als Signalvermittler
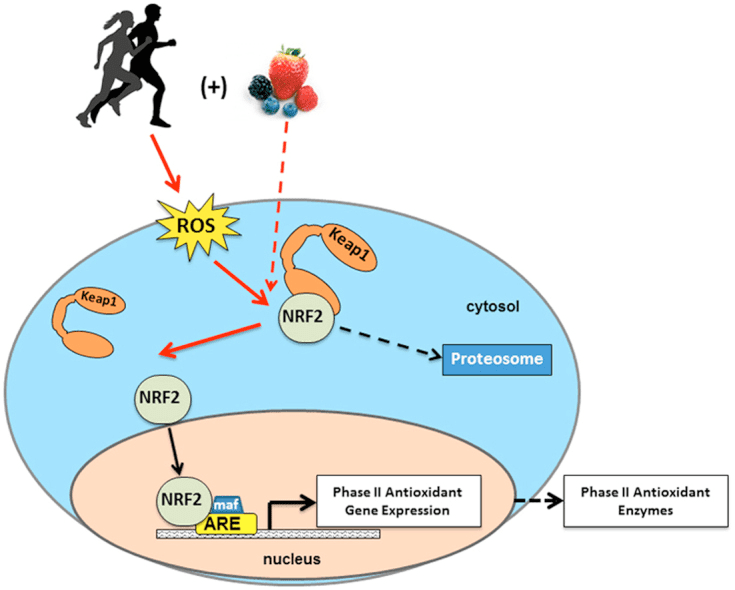
Obwohl sie energetisch reich sind, üben Ketonkörper provokative „nicht-kanonische“ Signalfunktionen in der zellulären Homöostase aus (Abb. 3) (Newman und Verdin, 2014; Rojas-Morales et al., 2016). ?OHB hemmt beispielsweise Klasse-I-HDACs, was die Histonacetylierung erhöht und dadurch die Expression von Genen induziert, die oxidativen Stress eindämmen (Shimazu et al., 2013). ?OHB selbst ist ein kovalenter Histon-Modifikator an Lysinresten in Lebern von nüchternen oder Streptozotocin-induzierten diabetischen Mäusen (Xie et al., 2016) (siehe auch unten, The integration of Keton body metadata, posttranslational modification, and cell physiology, und Ketonkörper, oxidativer Stress und Neuroprotektion).

?OHB ist auch ein Effektor über G-Protein-gekoppelte Rezeptoren. Durch unklare molekulare Mechanismen unterdrückt es die Aktivität des sympathischen Nervensystems und reduziert den Gesamtenergieverbrauch und die Herzfrequenz, indem es die Signalübertragung von kurzkettigen Fettsäuren durch den G-Protein-gekoppelten Rezeptor 41 (GPR41) hemmt (Kimura et al., 2011). Eine der am besten untersuchten Signalwirkungen von ?OHB erfolgt über GPR109A (auch bekannt als HCAR2), ein Mitglied der Hydrocarbonsäure-GPCR-Unterfamilie, das in Fettgeweben (weiß und braun) exprimiert wird (Tunaru et al., 2003) und in Immunzellen (Ahmed et al., 2009). ?OHB ist der einzige bekannte endogene Ligand des GPR109A-Rezeptors (EC50 ~770 µM), der durch d-?OHB, l-?OHB und Butyrat aktiviert wird, jedoch nicht durch AcAc (Taggart et al., 2005). Die hohe Konzentrationsschwelle für die GPR109A-Aktivierung wird durch Einhaltung einer ketogenen Diät, Hunger oder während einer Ketoazidose erreicht, was zu einer Hemmung der Fettgewebelipolyse führt. Die antilipolytische Wirkung von GPR109A erfolgt über die Hemmung der Adenylylcyclase und des verringerten cAMP, wodurch die hormonsensitive Triglyceridlipase gehemmt wird (Ahmed et al., 2009; Tunaru et al., 2003). Dadurch entsteht eine negative Rückkopplungsschleife, in der die Ketose die Ketogenese modulatorisch bremst, indem sie die Freisetzung nicht veresterter Fettsäuren aus Adipozyten verringert (Ahmed et al., 2009; Taggart et al., 2005), ein Effekt, der durch ausgeglichen werden kann der sympathische Antrieb, der die Lipolyse stimuliert. Niacin (Vitamin B3, Nikotinsäure) ist ein potenter (EC50 ~ 0.1 µM) Ligand für GRP109A, der seit Jahrzehnten bei Dyslipidämien wirksam eingesetzt wird (Benyo et al., 2005; Benyo et al., 2006; Fabbrini et al., 2010a; Lukasova et al., 2011; Tunaru et al., 2003). Während Niacin den reversen Cholesterintransport in Makrophagen verbessert und atherosklerotische Läsionen reduziert (Lukasova et al., 2011), sind die Auswirkungen von ?OHB auf atherosklerotische Läsionen unbekannt. Obwohl der GPR109A-Rezeptor eine schützende Rolle spielt und es faszinierende Verbindungen zwischen der ketogenen Ernährung bei Schlaganfall und neurodegenerativen Erkrankungen gibt (Fu et al., 2015; Rahman et al., 2014), wurde eine schützende Rolle von ?OHB über GPR109A in vivo nicht nachgewiesen .
Schließlich kann ?OHB Appetit und Sättigung beeinflussen. Eine Metaanalyse von Studien, die die Auswirkungen von ketogenen und sehr energiearmen Diäten maßen, kam zu dem Schluss, dass Teilnehmer, die diese Diäten zu sich nahmen, im Vergleich zu Kontrolldiäten ein höheres Sättigungsgefühl aufweisen (Gibson et al., 2015). Eine plausible Erklärung für diesen Effekt sind jedoch die zusätzlichen metabolischen oder hormonellen Elemente, die den Appetit modulieren könnten. Zum Beispiel zeigten Mäuse, die mit einer ketogenen Nagetierdiät gehalten wurden, trotz ähnlicher Kalorienaufnahme einen erhöhten Energieverbrauch im Vergleich zu Mäusen, die mit Kontrollfutter gefüttert wurden, und zirkulierendes Leptin oder Gene von Peptiden, die das Fressverhalten regulieren, wurden nicht verändert (Kennedy et al., 2007). Zu den vorgeschlagenen Mechanismen, die eine Appetitunterdrückung durch &agr;OHB nahelegen, gehören sowohl die Signalgebung als auch die Oxidation (Laeger et al., 2010). Hepatozytenspezifische Deletion des zirkadianen Rhythmusgens (Per2) und Chromatin-Immunpräzipitationsstudien zeigten, dass PER2 direkt das Cpt1a-Gen aktiviert und indirekt Hmgcs2 reguliert, was zu einer beeinträchtigten Ketose bei Per2-Knockout-Mäusen führt (Chavan et al., 2016). Diese Mäuse zeigten eine beeinträchtigte Nahrungserwartung, die teilweise durch systemische &bgr;OHB-Verabreichung wiederhergestellt wurde. Zukünftige Studien werden benötigt, um das Zentralnervensystem als direktes &bgr;OHB-Ziel zu bestätigen und ob für die beobachteten Effekte eine Ketonoxidation erforderlich ist oder ob ein anderer Signalmechanismus beteiligt ist. Andere Forscher haben die Möglichkeit einer lokalen Astrozyten-abgeleiteten Ketogenese im ventromedialen Hypothalamus als Regulator der Nahrungsaufnahme geltend gemacht, aber diese vorläufigen Beobachtungen werden auch von genetischen und flussbasierten Bewertungen profitieren (Le Foll et al., 2014). Der Zusammenhang zwischen Ketose und Nährstoffmangel bleibt von Interesse, da Hunger und Sättigung wichtige Elemente bei gescheiterten Gewichtsverlustversuchen sind.
Integration von Ketone-Body-Metabolismus, posttranslationaler Modifikation und Zellphysiologie
Ketonkörper tragen zu kompartimentierten Pools von Acetyl-CoA bei, einem Schlüsselintermediat, das eine wichtige Rolle im Zellstoffwechsel spielt (Pietrocola et al., 2015). Eine Rolle von Acetyl-CoA besteht darin, als Substrat für die Acetylierung zu dienen, eine enzymatisch katalysierte kovalente Histon-Modifikation (Choudhary et al., 2014; Dutta et al., 2016; Fan et al., 2015; Menzies et al., 2016 ). Eine große Anzahl von dynamisch acetylierten Mitochondrienproteinen, von denen viele durch nicht-enzymatische Mechanismen entstehen können, sind auch aus Computer-Proteomics-Studien hervorgegangen (Dittenhafer-Reed et al., 2015; Hebert et al., 2013; Rardin et al., 2013 Shimazu et al., 2010). Lysin-Deacetylasen verwenden einen Zink-Cofaktor (z. B. nucleocytosolische HDACs) oder NAD + als Co-Substrat (Sirtuins, SIRTs) (Choudhary et al., 2014; Menzies et al., 2016). Das Acetylproteom dient sowohl als Sensor als auch als Effektor des gesamten zellulären Acetyl-CoA-Pools, da physiologische und genetische Manipulationen jeweils zu nicht-enzymatischen globalen Variationen der Acetylierung führen (Weinert et al., 2014). Da intrazelluläre Metaboliten als Modulatoren der Acetylierung von Lysinresten dienen, ist es wichtig, die Rolle von Ketonkörpern zu berücksichtigen, deren Häufigkeit sehr dynamisch ist.
OHB ist ein epigenetischer Modifikator durch mindestens zwei Mechanismen. Erhöhte OHB-Spiegel, die durch Fasten, Kalorieneinschränkung, direkte Verabreichung oder längeres Training induziert werden, führen zu einer HDAC-Hemmung oder Histonacetyltransferase-Aktivierung (Marosi et al., 2016; Sleiman et al., 2016) oder zu oxidativem Stress (Shimazu et al., 2013). . Die OHB-Hemmung von HDAC3 könnte die Stoffwechselphysiologie von Neugeborenen regulieren (Rando et al., 2016). Unabhängig davon modifiziert & Dgr; OHB selbst Histon-Lysin-Reste direkt (Xie et al., 2016). Längeres Fasten oder Steptozotocin-induzierte diabetische Ketoazidose erhöhten die Histon-Hydroxybutyrylierung. Obwohl die Anzahl der Lysin-Hydroxybutyrylierungs- und Acetylierungsstellen vergleichbar war, wurde eine stöchiometrisch höhere Histon-Hydroxybutyrylierung als die Acetylierung beobachtet. Verschiedene Gene wurden durch Histon-Lysin-Hydroxybutyrylierung gegenüber Acetylierung oder Methylierung beeinflusst, was auf unterschiedliche Zellfunktionen hinweist. Ob die & agr; -Hydroxybutyrylierung spontan oder enzymatisch ist, ist nicht bekannt, erweitert jedoch den Bereich der Mechanismen durch Ketonkörper, die die Transkription dynamisch beeinflussen.
Essentielle Zellreprogrammierungsereignisse während der Kalorienrestriktion und des Nährstoffmangels können durch SIRT3- und SIRT5-abhängige mitochondriale Deacetylierung bzw. Desuccinylierung vermittelt werden, wodurch ketogene und ketolytische Proteine auf posttranslationaler Ebene in Leber- und extrahepatischen Geweben reguliert werden (Dittenhafer-Reed et al.,. 2015; Hebert et al., 2013; Rardin et al., 2013; Shimazu et al., 2010). Obwohl der stöchiometrische Vergleich besetzter Stellen nicht unbedingt direkt mit Verschiebungen des Stoffwechselflusses zusammenhängt, ist die mitochondriale Acetylierung dynamisch und kann eher durch die Acetyl-CoA-Konzentration oder den mitochondrialen pH-Wert als durch enzymatische Acetyltransferasen gesteuert werden (Wagner und Payne, 2013). Dass SIRT3 und SIRT5 die Aktivitäten von Ketonkörper-metabolisierenden Enzymen modulieren, wirft die Frage nach der wechselseitigen Rolle von Ketonen bei der Bildung des Acetylproteoms, Succinylproteoms und anderer dynamischer zellulärer Ziele auf. Da Variationen der Ketogenese die NAD + -Konzentrationen widerspiegeln, könnten die Ketonproduktion und -häufigkeit die Sirtuinaktivität regulieren und dadurch die gesamten Acetyl-CoA / Succinyl-CoA-Pools, das Acylproteom und damit die Mitochondrien- und Zellphysiologie beeinflussen. Die & agr; -Hydroxybutyrylierung von Enzym-Lysinresten könnte der zellulären Reprogrammierung eine weitere Schicht hinzufügen. In extrahepatischen Geweben kann die Oxidation des Ketonkörpers analoge Veränderungen der Zellhomöostase stimulieren. Während die Kompartimentierung von Acetyl-CoA-Pools stark reguliert ist und ein breites Spektrum zellulärer Veränderungen koordiniert, muss die Fähigkeit von Ketonkörpern, sowohl mitochondriale als auch cytoplasmatische Acetyl-CoA-Konzentrationen direkt zu formen, aufgeklärt werden (Chen et al., 2012; Corbet et al., 2016; Pougovkina et al., 2014; Schwer et al., 2009; Wellen und Thompson, 2012). Da die Acetyl-CoA-Konzentrationen streng reguliert sind und Acetyl-CoA membranundurchlässig ist, ist es wichtig, die Treibermechanismen zu berücksichtigen, die die Acetyl-CoA-Homöostase koordinieren, einschließlich der Produktionsraten und der terminalen Oxidation im TCA-Zyklus, der Umwandlung in Ketonkörper, Mitochondrien Ausfluss über Carnitin-Acetyltransferase (CrAT) oder Acetyl-CoA-Export nach Cytosol nach Umwandlung in Citrat und Freisetzung durch ATP-Citrat-Lyase (ACLY). Die Schlüsselrollen dieser letzteren Mechanismen im Zellacetylproteom und in der Homöostase erfordern ein abgestimmtes Verständnis der Rollen der Ketogenese und Ketonoxidation (Das et al., 2015; McDonnell et al., 2016; Moussaieff et al., 2015; Overmyer et al., 2015; Seiler et al., 2014; Seiler et al., 2015; Wellen et al., 2009; Wellen und Thompson, 2012). Konvergente Technologien in der Metabolomik und Acylproteomik bei der Festlegung genetisch manipulierter Modelle werden erforderlich sein, um Ziele und Ergebnisse festzulegen.
Anti- und entzündungshemmende Reaktionen auf Ketonkörper
Ketose- und Ketonkörper modulieren die Entzündung und die Funktion der Immunzellen, es wurden jedoch verschiedene und sogar diskrepante Mechanismen vorgeschlagen. Längerer Nährstoffmangel reduziert Entzündungen (Youm et al., 2015), aber die chronische Ketose von Typ-1-Diabetes ist ein entzündungsfördernder Zustand (Jain et al., 2002; Kanikarla-Marie und Jain, 2015; Kurepa et al., 2012) ). Mechanismusbasierte Signalrollen für & Dgr; OHB bei Entzündungen treten auf, weil viele Zellen des Immunsystems, einschließlich Makrophagen oder Monozyten, GPR109A reichlich exprimieren. Während & Dgr; OHB eine überwiegend entzündungshemmende Reaktion ausübt (Fu et al., 2014; Gambhir et al., 2012; Rahman et al., 2014; Youm et al., 2015), können hohe Konzentrationen von Ketonkörpern, insbesondere AcAc, auftreten eine entzündungsfördernde Reaktion auslösen (Jain et al., 2002; Kanikarla-Marie und Jain, 2015; Kurepa et al., 2012).
Die entzündungshemmenden Rollen von GPR109A-Liganden bei Atherosklerose, Fettleibigkeit, entzündlichen Darmerkrankungen, neurologischen Erkrankungen und Krebs wurden untersucht (Graff et al., 2016). Die GPR109A-Expression wird in RPE-Zellen von Diabetikermodellen, menschlichen Diabetikern (Gambhir et al., 2012) und in Mikroglia während der Neurodegeneration (Fu et al., 2014) erhöht. Die entzündungshemmenden Wirkungen von & Dgr; OHB werden durch die Überexpression von GPR109A in RPE-Zellen verstärkt und durch pharmakologische Hemmung oder genetisches Knockout von GPR109A aufgehoben (Gambhir et al., 2012). OHB und exogene Nikotinsäure (Taggart et al., 2005) verleihen TNF entzündungshemmende Wirkungen. oder LPS-induzierte Entzündung durch Verringern der Spiegel von proinflammatorischen Proteinen (iNOS, COX-2) oder sekretierten Zytokinen (TNF & agr;, IL-1 & agr;, IL-6, CCL2 / MCP-1), teilweise durch Hemmung von NF - B-Translokation (Fu et al., 2014; Gambhir et al., 2012). OHB verringert den ER-Stress und das NLRP3-Inflammasom und aktiviert die antioxidative Stressreaktion (Bae et al., 2016; Youm et al., 2015). Bei neurodegenerativen Entzündungen beinhaltet der GPR109A-abhängige? OHB-vermittelte Schutz jedoch keine Entzündungsmediatoren wie die Signalübertragung des MAPK-Signalwegs (z. B. ERK, JNK, S. 38) (Fu et al., 2014), sondern erfordert möglicherweise COX-1-abhängiges PGD2 Produktion (Rahman et al., 2014). Es ist faszinierend, dass der Makrophagen GPR109A eine neuroprotektive Wirkung in einem ischämischen Schlaganfallmodell ausüben muss (Rahman et al., 2014), aber die Fähigkeit von & Dgr; OHB, das NLRP3-Inflammasom in aus Knochenmark stammenden Makrophagen zu hemmen, ist GPR109A-unabhängig (Youm et al ., 2015). Obwohl die meisten Studien OHB mit entzündungshemmenden Wirkungen in Verbindung bringen, kann OHB entzündungsfördernd sein und die Marker für die Lipidperoxidation in Kälberhepatozyten erhöhen (Shi et al., 2014). Die entzündungshemmenden Wirkungen von & Dgr; OHB können daher vom Zelltyp, der & Dgr; OHB-Konzentration, der Expositionsdauer und der Anwesenheit oder Abwesenheit von Co-Modulatoren abhängen.
Im Gegensatz zu OHB kann AcAc entzündungsfördernde Signale aktivieren. Erhöhtes AcAc, insbesondere bei einer hohen Glukosekonzentration, verstärkt die Endothelzellverletzung durch einen von NADPH-Oxidase / oxidativem Stress abhängigen Mechanismus (Kanikarla-Marie und Jain, 2015). Hohe AcAc-Konzentrationen in der Nabelschnur diabetischer Mütter korrelierten mit einer höheren Proteinoxidationsrate und MCP-1-Konzentration (Kurepa et al., 2012). Ein hoher AcAc-Wert bei Diabetikern korrelierte mit TNF? Expression (Jain et al., 2002) und AcAc, jedoch nicht & Dgr; OHB, induzierten TNF & agr;, MCP-1-Expression, ROS-Akkumulation und verringerten cAMP-Spiegel in menschlichen U937-Monozytenzellen (Jain et al., 2002; Kurepa et al ., 2012).
Ketonkörperabhängige Signalphänomene werden häufig nur bei hohen Ketonkörperkonzentrationen (> 5 mM) ausgelöst, und bei vielen Studien werden Ketone durch unklare Mechanismen mit pro- oder entzündungshemmenden Wirkungen in Verbindung gebracht. Aufgrund der widersprüchlichen Wirkungen von & Dgr; OHB gegenüber AcAc auf die Entzündung und der Fähigkeit des AcAc / & Dgr; OHB-Verhältnisses, das mitochondriale Redoxpotential zu beeinflussen, vergleichen die besten Experimente zur Bewertung der Rolle von Ketonkörpern auf zelluläre Phänotypen die Wirkungen von AcAc und & dgr; OHB in unterschiedlichen Verhältnissen und in unterschiedlichen kumulativen Konzentrationen [z. B. (Saito et al., 2016)]. Schließlich kann AcAc kommerziell nur als Lithiumsalz oder als Ethylester gekauft werden, der vor der Verwendung eine Basenhydrolyse erfordert. Das Lithiumkation induziert unabhängig voneinander Signalübertragungskaskaden (Manji et al., 1995), und das AcAc-Anion ist labil. Schließlich können Studien mit racemischem d / l-OHB verwechselt werden, da nur das d-OHB-Stereoisomer zu AcAc oxidiert werden kann, d-OHB und l-OHB jedoch jeweils über GPR109A signalisieren und das NLRP3-Inflammasom hemmen können. und dienen als lipogene Substrate.
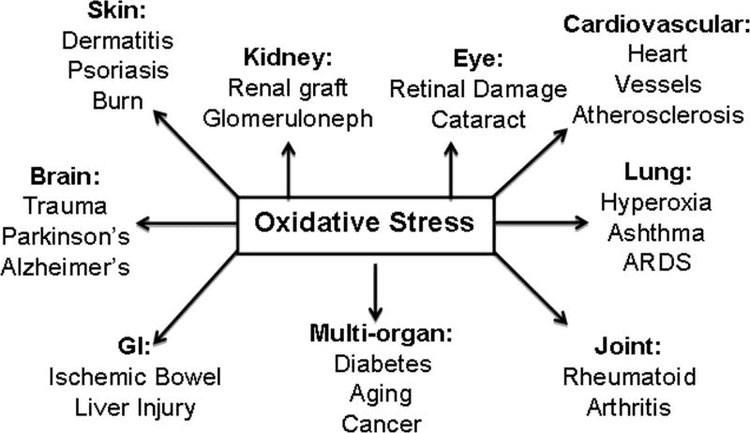
Ketonkörper, oxidativer Stress und Neuroprotektion
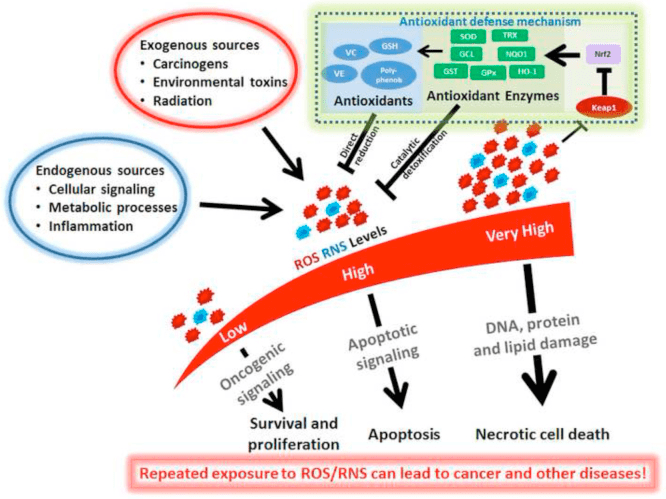
Oxidativer Stress wird typischerweise als ein Zustand definiert, in dem ROS aufgrund einer übermäßigen Produktion und/oder einer gestörten Elimination im Überschuss präsentiert werden. Die antioxidative und oxidative Stress abschwächende Rolle von Ketonkörpern wurde sowohl in vitro als auch in vivo umfassend beschrieben, insbesondere im Zusammenhang mit der Neuroprotektion. Da die meisten Neuronen nicht effektiv energiereiche Phosphate aus Fettsäuren erzeugen, aber Ketonkörper oxidieren, wenn Kohlenhydrate knapp sind, sind die neuroprotektiven Wirkungen von Ketonkörpern besonders wichtig (Cahill GF Jr, 2006; Edmond et al., 1987; Yang et al., 1987). In Modellen für oxidativen Stress deuten die BDH1-Induktion und die SCOT-Suppression darauf hin, dass der Stoffwechsel des Ketonkörpers umprogrammiert werden kann, um verschiedene Zellsignale, Redoxpotentiale oder metabolische Anforderungen aufrechtzuerhalten (Nagao et al., 2016; Tieu et al., 2003).
Ketonkörper verringern den Grad der Zellschädigung, -verletzung, -tod und verringern die Apoptose in Neuronen und Kardiomyozyten (Haces et al., 2008; Maalouf et al., 2007; Nagao et al., 2016; Tieu et al., 2003). Beschworene Mechanismen sind vielfältig und nicht immer linear mit der Konzentration verbunden. Niedrige millimolare Konzentrationen von (d oder l)-?OHB fangen ROS (Hydroxylanion) ab, während AcAc zahlreiche ROS-Spezies fängt, jedoch nur bei Konzentrationen, die den physiologischen Bereich überschreiten (IC50 20 mM) (Haces et al., 67) . Umgekehrt ist ein vorteilhafter Einfluss auf das Redoxpotential der Elektronentransportkette ein Mechanismus, der üblicherweise mit d-&ohgr;OHB in Verbindung gebracht wird. Während alle drei Ketonkörper (d/l-&ohgr;OHB und AcAc) den neuronalen Zelltod und die ROS-Akkumulation, ausgelöst durch die chemische Hemmung der Glykolyse, reduzierten, verhinderten nur d-&ohgr;OHB und AcAc den neuronalen ATP-Rückgang. Umgekehrt verhinderte in einem hypoglykämischen In-vivo-Modell (d oder l)-?OHB, aber nicht AcAc die Lipidperoxidation im Hippocampus (Haces et al., 2008; Maalouf et al., 2008; Marosi et al., 2007; Murphy, 2016 ; Tieu et al., 2009). In-vivo-Studien an Mäusen, die eine ketogene Diät erhielten (2003 % kcal Fett und 87 % Protein) zeigten eine neuroanatomische Variation der antioxidativen Kapazität (Ziegler et al., 13), wobei die tiefgreifendsten Veränderungen im Hippocampus beobachtet wurden, mit einem Anstieg von Glutathionperoxidase und Gesamt antioxidative Kapazitäten.
Ketogene Diät, Ketonester (siehe auch Therapeutische Verwendung von ketogener Diät und exogenen Ketonkörpern) oder OHB-Verabreichung üben Neuroprotektion in Modellen des ischämischen Schlaganfalls aus (Rahman et al., 2014); Parkinson-Krankheit (Tieu et al., 2003); Anfall durch Sauerstofftoxizität des zentralen Nervensystems (D'Agostino et al., 2013); epileptische Krämpfe (Yum et al., 2015); mitochondriale Enzephalomyopathie, Laktatazidose und Schlaganfall-ähnliches (MELAS)-Episoden-Syndrom (Frey et al., 2016) und Alzheimer-Krankheit (Cunnane und Crawford, 2003; Yin et al., 2016). Umgekehrt zeigte ein kürzlich veröffentlichter Bericht histopathologische Beweise für eine neurodegenerative Progression durch eine ketogene Ernährung in einem transgenen Mausmodell einer abnormalen mitochondrialen DNA-Reparatur, trotz einer Zunahme der mitochondrialen Biogenese und antioxidativer Signaturen (Lauritzen et al., 2016). Andere widersprüchliche Berichte deuten darauf hin, dass die Exposition gegenüber hohen Ketonkörperkonzentrationen oxidativen Stress hervorruft. Hohe ?OHB- oder AcAc-Dosen induzierten Stickoxid-Sekretion, Lipidperoxidation, reduzierte Expression von SOD, Glutathionperoxidase und Katalase in Kälberhepatozyten, während in Ratten-Hepatozyten die Induktion des MAPK-Signalwegs auf AcAc, aber nicht auf ?OHB zurückgeführt wurde (Abdelmegeed et al., 2004 ; Shi et al., 2014; Shi et al., 2016).
Zusammengenommen verbinden die meisten Berichte OHB mit der Abschwächung von oxidativem Stress, da seine Verabreichung die ROS / Superoxid-Produktion hemmt, die Lipidperoxidation und Proteinoxidation verhindert, die antioxidativen Proteinspiegel erhöht und die Mitochondrienatmung und die ATP-Produktion verbessert (Abdelmegeed et al., 2004; Haces et al., 2008; Jain et al., 1998; Jain et al., 2002; Kanikarla-Marie und Jain, 2015; Maalouf et al., 2007; Maalouf und Rho, 2008; Marosi et al., 2016; Tieu et al., 2003; Yin et al., 2016; Ziegler et al., 2003). Während AcAc direkter als & Dgr; OHB mit der Induktion von oxidativem Stress korreliert wurde, lassen sich diese Effekte nicht immer leicht von prospektiven proinflammatorischen Reaktionen trennen (Jain et al., 2002; Kanikarla-Marie und Jain, 2015; Kanikarla-Marie und Jain, 2016). Darüber hinaus ist es wichtig zu berücksichtigen, dass der offensichtliche antioxidative Nutzen, der durch pleiotrope ketogene Diäten verliehen wird, möglicherweise nicht von Ketonkörpern selbst übertragen wird und die durch Ketonkörper verliehene Neuroprotektion möglicherweise nicht vollständig auf oxidativen Stress zurückzuführen ist. Beispielsweise stimulierte OHB während eines Glukoseentzugs in einem Modell des Glukoseentzugs in kortikalen Neuronen den autophagischen Fluss und verhinderte die Akkumulation von Autophagosomen, was mit einem verringerten neuronalen Tod verbunden war (Camberos-Luna et al., 2016). d- & agr; OHB induziert auch die kanonischen Antioxidationsproteine FOXO3a, SOD, MnSOD und Katalase prospektiv durch HDAC-Hemmung (Nagao et al., 2016; Shimazu et al., 2013).
Alkoholfreie Fettlebererkrankung (NAFLD) und Ketone Body Metabolism
Adipositas-assoziierte NAFLD und nichtalkoholische Steatohepatitis (NASH) sind die häufigsten Ursachen für Lebererkrankungen in westlichen Ländern (Rinella und Sanyal, 2016), und NASH-induziertes Leberversagen ist einer der häufigsten Gründe für eine Lebertransplantation. Während eine übermäßige Lagerung von Triacylglycerinen in Hepatozyten> 5% des Lebergewichts (NAFL) allein keine degenerative Leberfunktion verursacht, korreliert das Fortschreiten der NAFLD beim Menschen mit der systemischen Insulinresistenz und dem erhöhten Risiko für Typ-2-Diabetes und kann zur Pathogenese von beitragen Herz-Kreislauf-Erkrankungen und chronische Nierenerkrankungen (Fabbrini et al., 2009; Targher et al., 2010; Targher und Byrne, 2013). Die pathogenen Mechanismen von NAFLD und NASH sind unvollständig verstanden, umfassen jedoch Anomalien des Hepatozytenstoffwechsels, der Hepatozytenautophagie und des Stresses des endoplasmatischen Retikulums, der Funktion der hepatischen Immunzellen, der Entzündung des Fettgewebes und systemischer Entzündungsmediatoren (Fabbrini et al., 2009; Masuoka und Chalasani, 2013) ; Targher et al., 2010; Yang et al., 2010). Störungen des Kohlenhydrat-, Lipid- und Aminosäurestoffwechsels treten beim Menschen und in Modellorganismen auf und tragen zu Fettleibigkeit, Diabetes und NAFLD bei [Übersicht in (Farese et al., 2012; Lin und Accili, 2011; Newgard, 2012; Samuel und Shulman, 2012; Sun und Lazar, 2013)]. Während bei NAFLD häufig Hepatozytenanomalien im zytoplasmatischen Lipidstoffwechsel beobachtet werden (Fabbrini et al., 2010b), ist die Rolle des mitochondrialen Stoffwechsels, der die oxidative Entsorgung von Fetten steuert, bei der NAFLD-Pathogenese weniger klar. Abnormalitäten des mitochondrialen Metabolismus treten in der NAFLD / NASH-Pathogenese auf und tragen dazu bei (Hyotylainen et al., 2016; Serviddio et al., 2011; Serviddio et al., 2008; Wei et al., 2008). Es gibt allgemeine (Felig et al., 1974; Iozzo et al., 2010; Koliaki et al., 2015; Satapati et al., 2015; Satapati et al., 2012; Sunny et al., 2011), aber nicht einheitliche ( Koliaki und Roden, 2013; Perry et al., 2016; Rector et al., 2010) sind sich einig, dass vor der Entwicklung von echtem NASH die mitochondriale Oxidation der Leber und insbesondere die Fettoxidation bei Fettleibigkeit und systemischer Insulinresistenz verstärkt wird und NAFLD. Es ist wahrscheinlich, dass mit fortschreitender NAFLD eine Heterogenität der oxidativen Kapazität selbst unter einzelnen Mitochondrien auftritt und letztendlich die oxidative Funktion beeinträchtigt wird (Koliaki et al., 2015; Rector et al., 2010; Satapati et al., 2008; Satapati et al ., 2012).
Die Ketogenese wird oft als Proxy für die Leberfettoxidation verwendet. Beeinträchtigungen der Ketogenese treten mit dem Fortschreiten der NAFLD in Tiermodellen und wahrscheinlich auch beim Menschen auf. Durch unvollständig definierte Mechanismen unterdrückt Hyperinsulinämie die Ketogenese und trägt möglicherweise im Vergleich zu schlanken Kontrollen zur Hypoketonämie bei (Bergman et al., 2007; Bickerton et al., 2008; Satapati et al., 2012; Soeters et al., 2009; Sunny et al. , 2011; Vice et al., 2005). Nichtsdestotrotz ist die Fähigkeit der zirkulierenden Ketonkörperkonzentrationen, NAFLD vorherzusagen, umstritten (M nnist et al., 2015; Sanyal et al., 2001). Robuste quantitative Magnetresonanzspektroskopiemethoden in Tiermodellen zeigten eine erhöhte Ketonumsatzrate bei mäßiger Insulinresistenz, aber verringerte Raten waren bei schwererer Insulinresistenz offensichtlich (Satapati et al., 2012; Sunny et al., 2010). Bei adipösen Menschen mit Fettleber ist die ketogene Rate normal (Bickerton et al., 2008; Sunny et al., 2011), und daher ist die Ketogeneserate im Verhältnis zur erhöhten Fettsäurebelastung in den Hepatozyten verringert. Folglich kann von &bgr;-Oxidation abgeleitetes Acetyl-CoA auf die terminale Oxidation im TCA-Zyklus gerichtet sein, was die terminale Oxidation, die Phosphoenolpyruvat-getriebene Gluconeogenese über Anaplerose/Kataplerose und oxidativen Stress erhöht. Acetyl-CoA wird möglicherweise auch als Citrat aus den Mitochondrien exportiert, einem Vorläufersubstrat für die Lipogenese (Abb. 4) (Satapati et al., 2015; Satapati et al., 2012; Solinas et al., 2015). Während die Ketogenese bei längerer Fettleibigkeit weniger auf Insulin oder Fasten anspricht (Satapati et al., 2012), bleiben die zugrunde liegenden Mechanismen und nachgelagerten Folgen davon unvollständig verstanden. Jüngste Hinweise deuten darauf hin, dass mTORC1 die Ketogenese auf eine Weise unterdrückt, die möglicherweise der Insulinsignalisierung nachgeschaltet ist (Kucejova et al., 2016), was mit den Beobachtungen übereinstimmt, dass mTORC1 die PPAR&bgr;-vermittelte Hmgcs2-Induktion hemmt (Sengupta et al., 2010) ( siehe auch Regulation von HMGCS2 und SCOT/OXCT1).

Vorläufige Beobachtungen unserer Gruppe deuten auf nachteilige Leberfolgen einer ketogenen Insuffizienz hin (Cotter et al., 2014). Um die Hypothese zu testen, dass eine beeinträchtigte Ketogenese selbst in kohlenhydratreichen und damit „nicht-ketogenen“ Zuständen zu einem abnormalen Glukosestoffwechsel beiträgt und eine Steatohepatitis provoziert, haben wir ein Mausmodell mit ausgeprägter ketogener Insuffizienz durch Verabreichung von Antisense-Oligonukleotiden (ASO) erstellt, die auf Hmgcs2. Der Verlust von HMGCS2 bei erwachsenen Mäusen, die mit normalem, fettarmem Futter gefüttert wurden, verursachte eine leichte Hyperglykämie und eine deutlich erhöhte Produktion von Hunderten von Lebermetaboliten, von denen eine Reihe stark auf eine Aktivierung der Lipogenese hindeutete. Die Fütterung von Mäusen mit ungenügender Ketogenese durch fettreiche Ernährung führte zu ausgedehnten Hepatozytenschäden und Entzündungen. Diese Ergebnisse unterstützen die zentralen Hypothesen, dass (i) die Ketogenese kein passiver Überlaufweg ist, sondern eher ein dynamischer Knoten in der hepatischen und integrierten physiologischen Homöostase, und (ii) eine umsichtige ketogene Augmentation zur Milderung von NAFLD/NASH und gestörtem hepatischen Glukosestoffwechsel eine Erforschung wert ist .
Wie könnte eine beeinträchtigte Ketogenese zu einer Leberschädigung und einer veränderten Glukosehomöostase beitragen? Die erste Überlegung ist, ob der Schuldige ein Mangel an ketogenem Fluss oder an Ketonen selbst ist. Ein kürzlich veröffentlichter Bericht legt nahe, dass Ketonkörper die durch oxidativen Stress verursachte Leberschädigung als Reaktion auf mehrfach ungesättigte n-3-Fettsäuren mildern können (Pawlak et al., 2015). Erinnern Sie sich daran, dass Ketonkörper aufgrund mangelnder SCOT-Expression in Hepatozyten nicht oxidiert werden, aber zur Lipogenese beitragen können und unabhängig von ihrer Oxidation eine Vielzahl von Signalfunktionen übernehmen (siehe auch Nichtoxidative metabolische Schicksale von Ketonkörpern und? OHB as ein Signalvermittler). Es ist auch möglich, dass von Hepatozyten abgeleitete Ketonkörper als Signal und / oder Metabolit für benachbarte Zelltypen innerhalb des Leberacinus dienen, einschließlich Sternzellen und Kupffer-Zellmakrophagen. Während die begrenzte verfügbare Literatur darauf hindeutet, dass Makrophagen nicht in der Lage sind, Ketonkörper zu oxidieren, wurde dies nur unter Verwendung klassischer Methoden und nur bei Peritonealmakrophagen gemessen (Newsholme et al., 1986; Newsholme et al., 1987), was darauf hinweist, dass eine erneute Die Beurteilung ist angesichts der reichlichen SCOT-Expression in aus Knochenmark stammenden Makrophagen angemessen (Youm et al., 2015).
Der ketogene Fluss von Hepatozyten kann auch zytoprotektiv sein. Während heilsame Mechanismen möglicherweise nicht per se von der Ketogenese abhängen, wurden ketogene Diäten mit niedrigem Kohlenhydratgehalt mit einer Verbesserung der NAFLD in Verbindung gebracht (Browning et al., 2011; Foster et al., 2010; Kani et al., 2014; Schugar und Crawford, 2012). . Unsere Beobachtungen deuten darauf hin, dass die Hepatozyten-Ketogenese den TCA-Zyklusfluss, den anaplerotischen Fluss, die Phosphoenolpyruvat-abgeleitete Glukoneogenese (Cotter et al., 2014) und sogar den Glykogenumsatz rückkoppeln und regulieren kann. Ketogene Beeinträchtigung führt Acetyl-CoA dazu, den TCA-Fluss zu erhöhen, was in der Leber mit einer erhöhten ROS-vermittelten Schädigung in Verbindung gebracht wurde (Satapati et al., 2015; Satapati et al., 2012); erzwingt die Ableitung von Kohlenstoff in de novo synthetisierte Lipidspezies, die sich als zytotoxisch erweisen könnten; und verhindert die Reoxidation von NADH zu NAD+ (Cotter et al., 2014) (Abb. 4). Zusammenfassend sind zukünftige Experimente erforderlich, um Mechanismen zu untersuchen, durch die eine relative ketogene Insuffizienz maladaptiv werden kann, zu Hyperglykämie beiträgt, eine Steatohepatitis hervorruft und ob diese Mechanismen bei humaner NAFLD/NASH wirksam sind. Da epidemiologische Hinweise darauf hindeuten, dass die Ketogenese während des Fortschreitens der Steatohepatitis beeinträchtigt ist (Embade et al., 2016; Marinou et al., 2011; M nnist et al., 2015; Pramfalk et al., 2015; Safaei et al., 2016) Therapien, die die hepatische Ketogenese steigern, könnten sich als heilsam erweisen (Degirolamo et al., 2016; Honda et al., 2016).
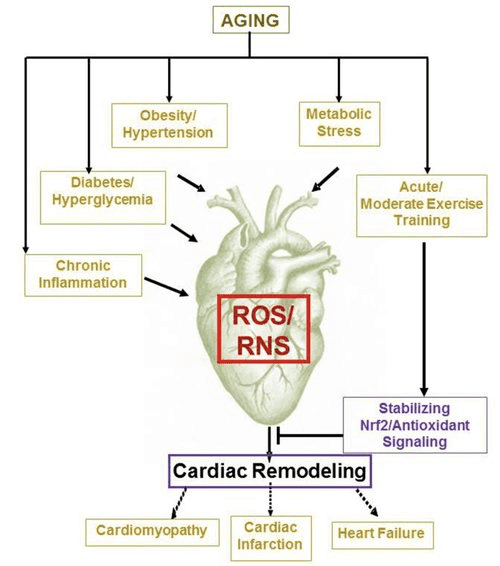
Ketonkörper und Herzinsuffizienz (HF)
Mit einer Stoffwechselrate von über 400 kcal/kg/Tag und einem Umsatz von 6 kg ATP/Tag ist das Herz das Organ mit dem höchsten Energieverbrauch und oxidativem Bedarf (Ashrafian et al., 35; Wang et al., 2007b). Der überwiegende Teil des myokardialen Energieumsatzes findet in den Mitochondrien statt, und 2010 % dieser Versorgung stammen aus der FAO. Das Herz ist unter normalen Bedingungen Allesfresser und flexibel, aber das sich pathologisch umbauende Herz (z. B. aufgrund von Bluthochdruck oder Myokardinfarkt) und das diabetische Herz werden jeweils metabolisch unflexibel (Balasse und Fery, 70; BING, 1989; Fukao et al., 1954). ; Lopaschuk et al., 2004; Taegtmeyer et al., 2010; Taegtmeyer et al., 1980; Young et al., 2002). Tatsächlich provozieren genetisch programmierte Anomalien des kardialen Brennstoffstoffwechsels in Mausmodellen eine Kardiomyopathie (Carley et al., 2002; Neubauer, 2014). Unter physiologischen Bedingungen oxidieren normale Herzen Ketonkörper proportional zu ihrer Abgabe, auf Kosten der Fettsäure- und Glukoseoxidation, und das Myokard ist der größte Ketonkörperverbraucher pro Masseneinheit (BING, 2007; Crawford et al., 1954; GARLAND et al.) ., 2009; Hasselbaink et al., 1962; Jeffrey et al., 2003; Pelletier et al., 1995; Tardif et al., 2007; Yan et al., 2001). Im Vergleich zur Fettsäureoxidation sind Ketonkörper energetisch effizienter und liefern mehr Energie für die ATP-Synthese pro eingesetztem Sauerstoffmolekül (P/O-Verhältnis) (Kashiwaya et al., 2009; Sato et al., 2010; Veech, 1995) . Die Ketonkörperoxidation liefert auch potenziell höhere Energie als FAO, wodurch Ubichinon oxidiert bleibt, was die Redoxspanne in der Elektronentransportkette erhöht und mehr Energie zur Synthese von ATP zur Verfügung stellt (Sato et al., 2004; Veech, 1995). Die Oxidation von Ketonkörpern kann auch die ROS-Produktion und damit den oxidativen Stress einschränken (Veech, 2004).
Vorläufige Interventions- und Beobachtungsstudien weisen auf eine potenzielle heilsame Rolle von Ketonkörpern im Herzen hin. Im Zusammenhang mit experimenteller Ischämie / Reperfusionsverletzung vermittelten Ketonkörper potentiell kardioprotektive Wirkungen (Al-Zaid et al., 2007; Wang et al., 2008), möglicherweise aufgrund der Zunahme der mitochondrialen Abundanz im Herzen oder der Hochregulierung der entscheidenden oxidativen Phosphorylierung Mediatoren (Snorek et al., 2012; Zou et al., 2002). Kürzlich durchgeführte Studien zeigen, dass die Nutzung von Ketonkörpern in ausgefallenen Herzen von Mäusen (Aubert et al., 2016) und Menschen (Bedi et al., 2016) erhöht ist, was frühere Beobachtungen beim Menschen unterstützt (BING, 1954; Fukao et al., 2000; Janardhan et al., 2011; Longo et al., 2004; Rudolph und Schinz, 1973; Tildon und Cornblath, 1972). Die Konzentration von zirkulierenden Ketonkörpern ist bei Herzinsuffizienzpatienten im direkten Verhältnis zum Füllungsdruck und Beobachtungen, deren Mechanismus und Bedeutung unbekannt sind, erhöht (Kupari et al., 1995; Lommi et al., 1996; Lommi et al., 1997; Neely et al (1972), aber Mäuse mit selektivem SCOT-Mangel in Kardiomyozyten zeigen ein beschleunigtes pathologisches ventrikuläres Remodeling und ROS-Signaturen als Reaktion auf eine chirurgisch induzierte Drucküberlastungsverletzung (Schugar et al., 2014).
Kürzlich interessante Beobachtungen in der Diabetestherapie haben einen potenziellen Zusammenhang zwischen dem Metabolismus des Myokardketons und dem pathologischen ventrikulären Remodeling gezeigt (Abb. 5). Die Hemmung des renalen proximalen tubulären Natrium / Glucose-Co-Transporters 2 (SGLT2i) erhöht die zirkulierenden Ketonkörperkonzentrationen beim Menschen (Ferrannini et al., 2016a; Inagaki et al., 2015) und Mäuse (Suzuki et al., 2014) hepatische Ketogenese (Ferrannini et al., 2014; Ferrannini et al., 2016a; Katz und Leiter, 2015; Mudaliar et al., 2015). Es ist auffallend, dass mindestens eines dieser Mittel die HF-Hospitalisierung (z. B. durch die EMPA-REG OUTCOME-Studie) verringerte und die kardiovaskuläre Mortalität verbesserte (Fitchett et al., 2016; Sonesson et al., 2016; Wu et al., 2016a Zinman et al., 2015). Während die treibenden Mechanismen hinter günstigen HF-Ergebnissen für die Verknüpfung von SGLT2i weiterhin aktiv diskutiert werden, ist der Überlebensvorteil wahrscheinlich multifaktoriell und umfasst voraussichtlich Ketose, aber auch heilsame Auswirkungen auf Gewicht, Blutdruck, Glukose- und Harnsäurespiegel, arterielle Steifheit, das sympathische Nervensystem und osmotisch Diurese / reduziertes Plasmavolumen und erhöhter Hämatokrit (Raz und Cahn, 2016; Vallon und Thomson, 2016). Zusammengefasst bleibt der Gedanke, dass eine therapeutisch ansteigende Ketonämie entweder bei HF-Patienten oder bei Patienten mit einem hohen Risiko für die Entwicklung von HF besteht, umstritten, wird jedoch in präklinischen und klinischen Studien aktiv untersucht (Ferrannini et al., 2016b; Kolwicz et al. 2016; Lopaschuk und Verma, 2016; Mudaliar et al., 2016; Taegtmeyer, 2016).

Ketonkörper in der Krebsbiologie
Verbindungen zwischen Ketonkörpern und Krebs entwickeln sich rasch, aber Studien sowohl an Tiermodellen als auch am Menschen haben zu verschiedenen Schlussfolgerungen geführt. Da der Ketonstoffwechsel dynamisch ist und auf den Nährstoffzustand reagiert, ist es aufgrund des Potenzials für präzise geführte Ernährungstherapien verlockend, biologische Verbindungen zu Krebs zu suchen. Krebszellen durchlaufen eine metabolische Reprogrammierung, um eine schnelle Zellproliferation und -wachstum aufrechtzuerhalten (DeNicola und Cantley, 2015; Pavlova und Thompson, 2016). Der klassische Warburg-Effekt im Krebszellstoffwechsel beruht auf der dominierenden Rolle der Glykolyse und der Milchsäuregärung bei der Energieübertragung und zum Ausgleich einer geringeren Abhängigkeit von oxidativer Phosphorylierung und einer eingeschränkten Atmung der Mitochondrien (De Feyter et al., 2016; Grabacka et al., 2016; Kang et al., 2015; Poff et al., 2014; Shukla et al., 2014). Glucose-Kohlenstoff wird hauptsächlich durch die Glykolyse, den Pentosephosphatweg und die Lipogenese geleitet, die zusammen die für die Tumorbiomasse-Expansion erforderlichen Intermediate bereitstellen (Grabacka et al., 2016; Shukla et al., 2014; Yoshii et al., 2015). Die Anpassung von Krebszellen an den Glukosemangel erfolgt durch die Nutzung alternativer Kraftstoffquellen, einschließlich Acetat, Glutamin und Aspartat (Jaworski et al., 2016; Sullivan et al., 2015). Beispielsweise zeigt ein eingeschränkter Zugang zu Pyruvat die Fähigkeit von Krebszellen, Glutamin durch Carboxylierung in Acetyl-CoA umzuwandeln, wodurch sowohl der energetische als auch der anabolische Bedarf erhalten bleibt (Yang et al., 2014). Eine interessante Anpassung von Krebszellen ist die Verwendung von Acetat als Brennstoff (Comerford et al., 2014; Jaworski et al., 2016; Mashimo et al., 2014; Wright und Simone, 2016; Yoshii et al., 2015). Acetat ist auch ein Substrat für die Lipogenese, die für die Tumorzellproliferation kritisch ist, und der Gewinn dieses lipogenen Kanals ist mit einem kürzeren Überleben des Patienten und einer größeren Tumorlast verbunden (Comerford et al., 2014; Mashimo et al., 2014; Yoshii et al 2015).
Nicht-Krebszellen verlagern ihre Energiequelle während des Glukoseentzugs leicht von Glukose auf Ketonkörper. Diese Plastizität kann bei Krebszelltypen unterschiedlicher sein, aber in vivo implantierte Hirntumore oxidierten [2,4-13C2]-&ohgr;OHB in einem ähnlichen Ausmaß wie das umgebende Hirngewebe (De Feyter et al., 2016). „Umgekehrter Warburg-Effekt“ oder „Zwei-Kompartiment-Tumormetabolismus“-Modelle gehen davon aus, dass Krebszellen die OHB-Produktion in benachbarten Fibroblasten induzieren und den Energiebedarf der Tumorzelle decken (Bonuccelli et al., 2010; Martinez-Outschoorn et al., 2012) . In der Leber stimmt eine Verschiebung der Hepatozyten von der Ketogenese zur Ketonoxidation in hepatozellulären Karzinomzellen (Hepatom) mit der Aktivierung von BDH1- und SCOT-Aktivitäten überein, die in zwei Hepatomzelllinien beobachtet wurden (Zhang et al., 1989). Tatsächlich exprimieren Hepatomzellen OXCT1 und BDH1 und oxidieren Ketone, aber nur bei Serummangel (Huang et al., 2016). Alternativ wurde auch die Ketogenese von Tumorzellen vorgeschlagen. Dynamische Verschiebungen der ketogenen Genexpression werden während der kanzerösen Transformation des Kolonepithels, eines Zelltyps, der normalerweise HMGCS2 exprimiert, gezeigt, und ein kürzlich veröffentlichter Bericht deutet darauf hin, dass HMGCS2 ein prognostischer Marker für eine schlechte Prognose bei kolorektalen und Plattenepithelkarzinomen sein könnte (Camarero et al., 2006; Chen et al., 2016). Ob diese Assoziation eine Ketogenese oder eine Schwarzarbeitsfunktion von HMGCS2 erfordert oder beinhaltet, muss noch geklärt werden. Umgekehrt ist die scheinbare &bgr;OHB-Produktion durch Melanom- und Glioblastomzellen, stimuliert durch das PPAR&agr; Agonist Fenofibrat, wurde mit einem Wachstumsstillstand in Verbindung gebracht (Grabacka et al., 2016). Weitere Studien sind erforderlich, um die Rolle der HMGCS2/SCOT-Expression, Ketogenese und Ketonoxidation in Krebszellen zu charakterisieren.
Über den Bereich des Kraftstoffstoffwechsels hinaus wurden Ketone kürzlich über einen Signalmechanismus in die Krebszellbiologie einbezogen. Die Analyse des BRAF-V600E + -Melanoms zeigte eine OCT1-abhängige Induktion von HMGCL in einer onkogenen BRAF-abhängigen Weise (Kang et al., 2015). Die HMGCL-Augmentation korrelierte mit einer höheren zellulären AcAc-Konzentration, was wiederum die BRAFV600E-MEK1-Interaktion verstärkte und die MEK-ERK-Signalübertragung in einer Feed-Forward-Schleife verstärkte, die die Proliferation und das Wachstum von Tumorzellen antreibt. Diese Beobachtungen werfen die faszinierende Frage der prospektiven extrahepatischen Ketogenese auf, die dann einen Signalmechanismus unterstützt (siehe auch OHB als Signalvermittler und Kontroversen in der extrahepatischen Ketogenese). Es ist auch wichtig, unabhängige Wirkungen von AcAc, d-OHB und l-OHB auf den Krebsstoffwechsel zu berücksichtigen, und wenn HMGCL in Betracht gezogen wird, kann auch der Leucinkatabolismus gestört sein.
Die Auswirkungen ketogener Diäten (siehe auch Therapeutischer Einsatz von ketogener Diät und exogenen Ketonkörpern) in Krebstiermodellen sind vielfältig (De Feyter et al., 2016; Klement et al., 2016; Meidenbauer et al., 2015; Poff et al ., 2014; Seyfried et al., 2011; Shukla et al., 2014). Während epidemiologische Assoziationen zwischen Fettleibigkeit, Krebs und ketogenen Diäten diskutiert werden (Liskiewicz et al., 2016; Wright und Simone, 2016), deutete eine Metaanalyse mit ketogenen Diäten in Tiermodellen und in Humanstudien einen heilsamen Einfluss auf das Überleben an, mit Vorteile, die prospektiv mit dem Ausmaß der Ketose, dem Zeitpunkt der Diäteinleitung und der Tumorlokalisation verbunden sind (Klement et al., 2016; Woolf et al., 2016). Die Behandlung von Bauchspeicheldrüsenkrebszellen mit Ketonkörpern (d-?OHB oder AcAc) hemmte das Wachstum, die Proliferation und die Glykolyse, und eine ketogene Ernährung (81% kcal Fett, 18% Protein, 1% Kohlenhydrat) reduzierte in vivo Tumorgewicht, Glykämie und erhöhtes Muskel- und Körpergewicht bei Tieren mit implantiertem Krebs (Shukla et al., 2014). Ähnliche Ergebnisse wurden unter Verwendung eines metastasierten Glioblastom-Zellmodells bei Mäusen beobachtet, die eine Keton-Supplementierung in der Nahrung erhielten (Poff et al., 2014). Umgekehrt erhöhte eine ketogene Diät (91 % kcal Fett, 9 % Protein) die zirkulierende ?OHB-Konzentration und verringerte die Glykämie, hatte jedoch keinen Einfluss auf das Tumorvolumen oder die Überlebensdauer bei gliomtragenden Ratten (De Feyter et al., 2016). Ein Glukoseketonindex wurde als klinischer Indikator vorgeschlagen, der das metabolische Management der ketogenen ernährungsinduzierten Hirntumortherapie bei Menschen und Mäusen verbessert (Meidenbauer et al., 2015). Zusammengenommen sind die Rollen des Ketonkörpermetabolismus und der Ketonkörper in der Krebsbiologie verlockend, da sie jeweils handhabbare therapeutische Optionen darstellen, aber grundlegende Aspekte müssen noch geklärt werden, wobei klare Einflüsse aus einer Matrix von Variablen hervorgehen, einschließlich (i) Unterschieden zwischen exogenen Ketonen Körper versus ketogene Ernährung, (ii) Krebszelltyp, genomische Polymorphismen, Grad und Stadium; und (iii) Zeitpunkt und Dauer der Exposition gegenüber dem ketotischen Zustand.

Ketogenese wird durch Ketonkörper durch den Abbau von Fettsäuren und ketogenen Aminosäuren erzeugt. Dieser biochemische Prozess versorgt verschiedene Organe, insbesondere das Gehirn, unter Fasten als Reaktion auf eine Nichtverfügbarkeit von Blutglukose mit Energie. Ketonkörper werden hauptsächlich in den Mitochondrien von Leberzellen produziert. Während andere Zellen zur Ketogenese befähigt sind, sind sie dabei nicht so wirksam wie Leberzellen. Da die Ketogenese in den Mitochondrien vorkommt, werden ihre Prozesse unabhängig voneinander reguliert. Dr. Alex Jiménez DC, CCST Insight
Therapeutische Anwendung der ketogenen Diät und exogener Ketonkörper
Die Anwendung von ketogenen Diäten und Ketonkörpern als therapeutische Instrumente ist auch in nicht-krebsartigen Kontexten wie Fettleibigkeit und NAFLD/NASH aufgetreten (Browning et al., 2011; Foster et al., 2010; Schugar und Crawford, 2012); Herzinsuffizienz (Huynh, 2016; Kolwicz et al., 2016; Taegtmeyer, 2016); neurologische und neurodegenerative Erkrankungen (Martin et al., 2016; McNally und Hartman, 2012; Rho, 2015; Rogawski et al., 2016; Yang und Cheng, 2010; Yao et al., 2011); angeborene Stoffwechselstörungen (Scholl-Bärgi et al, 2015); und Trainingsleistung (Cox et al., 2016). Die Wirksamkeit von ketogenen Diäten wurde besonders bei der Therapie von epileptischen Anfällen, insbesondere bei arzneimittelresistenten Patienten, geschätzt. Die meisten Studien haben ketogene Diäten bei pädiatrischen Patienten evaluiert und zeigen eine bis zu 50 %ige Verringerung der Anfallshäufigkeit nach 3 Monaten mit verbesserter Wirksamkeit bei ausgewählten Syndromen (Wu et al., 2016b). Bei Epilepsie bei Erwachsenen sind die Erfahrungen begrenzter, aber eine ähnliche Reduktion ist offensichtlich, mit einem besseren Ansprechen bei Patienten mit symptomatischer generalisierter Epilepsie (Nei et al., 2014). Die zugrunde liegenden antikonvulsiven Mechanismen bleiben unklar, obwohl postulierte Hypothesen eine reduzierte Glukoseverwertung/Glykolyse, einen umprogrammierten Glutamattransport, einen indirekten Einfluss auf den ATP-sensitiven Kaliumkanal oder den Adenosin-A1-Rezeptor, eine Veränderung der Isoformexpression des Natriumkanals oder Auswirkungen auf zirkulierende Hormone einschließlich Leptin ( Lambrechts et al., 2016; Lin et al., 2017; Lutas und Yellen, 2013). Es bleibt unklar, ob die antikonvulsive Wirkung hauptsächlich auf Ketonkörper zurückzuführen ist oder auf die kaskadenartigen metabolischen Folgen einer kohlenhydratarmen Ernährung. Dennoch scheinen Ketonester (siehe unten) die Anfallsschwelle in Tiermodellen für provozierte Anfälle zu erhöhen (Ciarlone et al., 2016; D'Agostino et al., 2013; Viggiano et al., 2015).
Atkins-artige und ketogene, kohlenhydratarme Diäten werden häufig als unangenehm empfunden und können Verstopfung, Hyperurikämie, Hypocalcämie, Hypomagnesiämie, Nephrolithiasis, Ketoazidose, Hyperglykämie und zirkulierende Cholesterin- und freie Fettsäurekonzentrationen verursachen (Bisschop et al., 2001) Kossoff und Hartman, 2012; Kwiterovich et al., 2003; Suzuki et al., 2002). Aus diesen Gründen stellt die langfristige Einhaltung eine Herausforderung dar. Nagetierstudien verwenden üblicherweise eine ausgeprägte Makronährstoffverteilung (94% kcal Fett, 1% kcal Kohlenhydrat, 5% kcal Protein, Bio-Serv F3666), die eine robuste Ketose hervorruft. Eine Erhöhung des Proteingehaltes sogar auf 10% kcal verringert jedoch die Ketose wesentlich, und die 5% kcal-Proteinrestriktion führt zu verwirrenden metabolischen und physiologischen Wirkungen. Diese Diätformulierung ist auch an Cholin erschöpft, eine weitere Variable, die die Empfindlichkeit gegenüber Leberschäden und sogar die Ketogenese beeinflusst (Garbow et al., 2011; Jornayvaz et al., 2010; Kennedy et al., 2007; Pissios et al., 2013; Schugar et al., 2013). Die Auswirkungen des Langzeitkonsums ketogener Diäten bei Mäusen bleiben unvollständig, aber kürzlich durchgeführte Studien an Mäusen zeigten ein normales Überleben und das Fehlen von Leberschädigungsmarkern bei Mäusen auf ketogenen Diäten während ihrer gesamten Lebensdauer, obwohl Aminosäuremetabolismus, Energieverbrauch und Insulinsignalisierung wurden deutlich umprogrammiert (Douris et al., 2015).
Mechanismen, die die Ketose durch alternative Mechanismen zu ketogenen Diäten steigern, umfassen die Verwendung einnehmbarer Ketonkörper-Vorläufer. Die Verabreichung von exogenen Ketonkörpern könnte einen einzigartigen physiologischen Zustand schaffen, der in der normalen Physiologie nicht anzutreffen ist, da zirkulierende Glukose- und Insulinkonzentrationen relativ normal sind, während Zellen Glukoseaufnahme und -verwendung ersparen. Ketonkörper selbst haben kurze Halbwertszeiten, und die Einnahme oder Infusion von Natrium-OHB-Salz zur Erzielung einer therapeutischen Ketose führt zu einer ungünstigen Natriumbelastung. R / S-1,3-Butandiol ist ein ungiftiger Dialkohol, der in der Leber leicht zu d / l-OHB oxidiert wird (Desrochers et al., 1992). In verschiedenen experimentellen Zusammenhängen wurde diese Dosis Mäusen oder Ratten sieben Wochen lang täglich verabreicht, was zirkulierende OHB-Konzentrationen von bis zu 5 mM innerhalb von 2 Stunden nach der Verabreichung ergab, was für mindestens weitere 3 Stunden stabil ist (D '). Agostino et al., 2013). Bei Nagern, denen R / S-1,3-Butandiol (Carpenter und Grossman, 1983) verabreicht wurde, wurde eine teilweise Unterdrückung der Nahrungsaufnahme beobachtet. Zusätzlich drei chemisch unterschiedliche Ketonester (KEs), (i) Monoester von R-1,3-Butandiol und d-OHB (R-3-Hydroxybutyl-R-OHB); (ii) Glyceryltris-OHB; und (iii) R, S-1,3-Butandiolacetoacetat-Diester, wurden ebenfalls ausführlich untersucht (Brunengraber, 1997; Clarke et al., 2012a; Clarke et al., 2012b; Desrochers et al., 1995a; Desrochers et al ., 1995b; Kashiwaya et al., 2010). Ein inhärenter Vorteil des ersteren besteht darin, dass 2 Mol physiologisches d- & agr; OHB pro Mol KE nach Esterasehydrolyse im Darm oder in der Leber produziert werden. Sicherheit, Pharmakokinetik und Verträglichkeit wurden am ausführlichsten bei Menschen untersucht, die R-3-Hydroxybutyl-R-OHB in Dosen von bis zu 714 mg / kg einnahmen, was zirkulierende d-OHB-Konzentrationen von bis zu 6 mM ergab (Clarke et al., 2012a; Cox et al., 2016; Kemper et al., 2015; Shivva et al., 2016). Bei Nagetieren verringert diese KE die Kalorienaufnahme und das Gesamtcholesterin im Plasma, stimuliert braunes Fettgewebe und verbessert die Insulinresistenz (Kashiwaya et al., 2010; Kemper et al., 2015; Veech, 2013). Jüngste Erkenntnisse deuten darauf hin, dass während des Trainings bei trainierten Sportlern die Aufnahme von R-3-Hydroxybutyl-R- & OHgr; OHB die Glykolyse der Skelettmuskulatur und die Plasma-Laktat-Konzentrationen verringerte, die intramuskuläre Triacylglycerin-Oxidation erhöhte und den Glykogengehalt der Muskeln beibehielt, selbst wenn die kohlenhydratstimulierte Insulinsekretion gleichzeitig aufgenommen wurde ( Cox et al., 2016). Diese faszinierenden Ergebnisse müssen weiterentwickelt werden, da die Verbesserung der Leistung bei Ausdauerübungen vor allem von einer robusten Reaktion auf die KE bei 2 / 8-Patienten getrieben wurde. Nichtsdestotrotz unterstützen diese Ergebnisse klassische Studien, die auf eine Bevorzugung der Ketonoxidation gegenüber anderen Substraten hinweisen (GARLAND ua, 1962; Hasselbaink ua, 2003; Stanley ua, 2003; Valente-Silva ua, 2015). einschließlich während des Trainings, und dass ausgebildete Sportler besser darauf vorbereitet sein können, Ketone zu verwenden (Johnson et al., 1969a; Johnson und Walton, 1972; Winder et al., 1974; Winder et al., 1975). Schließlich müssen noch die Mechanismen ermittelt werden, die eine verbesserte Trainingsleistung nach einer gleichen Kalorienzufuhr (differenziell verteilt auf Makronährstoffe) und gleichen Sauerstoffverbrauchsraten unterstützen.
Zukunftsperspektive
Einst weitgehend als Überlaufweg stigmatisiert, der toxische Emissionen aus der Fettverbrennung in kohlenhydratarmen Zuständen akkumulieren kann (das „ketotoxische“ Paradigma), stützen neuere Beobachtungen die Vorstellung, dass der Stoffwechsel des Ketonkörpers selbst in kohlenhydratreichen Zuständen eine heilsame Rolle spielt und ein „Ketohormetikum“ eröffnet Hypothese. Während die einfachen ernährungsphysiologischen und pharmakologischen Ansätze zur Manipulation des Ketonstoffwechsels es zu einem attraktiven therapeutischen Ziel machen, bleiben aggressiv gestellte, aber umsichtige Experimente sowohl in den Grundlagen- als auch in den translationalen Forschungslabors. Ungedeckte Bedürfnisse sind in den Bereichen der Definition der Rolle der Nutzung des Ketonstoffwechsels bei Herzinsuffizienz, Fettleibigkeit, NAFLD/NASH, Typ-2-Diabetes und Krebs aufgetreten. Der Umfang und die Auswirkungen „nicht-kanonischer“ Signalfunktionen von Ketonkörpern, einschließlich der Regulierung von PTMs, die wahrscheinlich in Stoffwechsel- und Signalwege zurück- und weiterleiten, erfordern eine tiefere Untersuchung. Schließlich könnte die extrahepatische Ketogenese faszinierende parakrine und autokrine Signalmechanismen und Möglichkeiten eröffnen, den Kom-Stoffwechsel innerhalb des Nervensystems und von Tumoren zu beeinflussen, um therapeutische Ziele zu erreichen.
Anerkennungen
Ncbi.nlm.nih.gov/pmc/articles/PMC5313038/
Fußnoten
Zusammenfassend lässt sich sagen, dass Ketonkörper von der Leber gebildet werden, um als Energiequelle verwendet zu werden, wenn im menschlichen Körper nicht genügend Glukose verfügbar ist. Ketogenese tritt bei niedrigen Glukosespiegeln im Blut auf, insbesondere nachdem andere zelluläre Kohlenhydratspeicher erschöpft sind. Der Zweck des obigen Artikels war es, die mehrdimensionalen Rollen von Ketonkörpern im Kraftstoffstoffwechsel, bei der Signalübertragung und bei Therapeutika zu diskutieren. Der Umfang unserer Informationen beschränkt sich auf Chiropraktik und Wirbelsäulengesundheit. Um das Thema zu besprechen, wenden Sie sich bitte an Dr. Jimenez oder kontaktieren Sie uns unter 915-850-0900 .
Kuratiert von Dr. Alex Jimenez
Referenziert von: Ncbi.nlm.nih.gov/pmc/articles/PMC5313038/

Zusätzliche Themendiskussion: Akute Rückenschmerzen
RückenschmerzenDies ist eine der häufigsten Ursachen für Behinderungen und verpasste Arbeitstage weltweit. Rückenschmerzen sind der zweithäufigste Grund für Arztbesuche, der nur durch Infektionen der oberen Atemwege übertroffen wird. Ungefähr 80 Prozent der Bevölkerung leiden mindestens einmal im Laufe ihres Lebens an Rückenschmerzen. Die Wirbelsäule ist eine komplexe Struktur, die unter anderem aus Knochen, Gelenken, Bändern und Muskeln besteht. Verletzungen und / oder verschärfte Zustände, wie zBandscheibenvorfall, kann schließlich zu Symptomen von Rückenschmerzen führen. Sportverletzungen oder Autounfallverletzungen sind oft die häufigste Ursache für Rückenschmerzen, doch manchmal können einfachste Bewegungen schmerzhafte Folgen haben. Glücklicherweise können alternative Behandlungsoptionen wie Chiropraktik Rückenschmerzen durch Wirbelsäulenanpassungen und manuelle Manipulationen lindern und letztendlich die Schmerzlinderung verbessern.

EXTRA EXTRA | WICHTIGES THEMA: Empfohlen El Paso, TX Chiropraktiker
***